Ewing Sarcoma: Comprehensive Pathology, Diagnosis, and Surgical Management
Key Takeaway
Ewing sarcoma is a highly aggressive, small round blue cell tumor primarily affecting children and young adults. It typically arises in the metadiaphysis of long bones or the flat bones of the pelvis. Multidisciplinary management is mandatory, combining neoadjuvant chemotherapy with definitive local control via wide surgical resection or radiotherapy. This guide details the diagnostic workup, molecular pathology, and step-by-step surgical strategies for optimizing oncologic and functional outcomes.
Introduction and Epidemiology
Ewing sarcoma represents a highly aggressive, high-grade primary malignancy of bone and soft tissue. It is the fourth most common primary malignancy of bone overall; however, its demographic distribution makes it a critical diagnostic consideration in pediatric and young adult populations. It is the second most common primary bone malignancy (following osteosarcoma) in patients younger than 30 years of age, and it stands as the most common primary bone sarcoma in children younger than 10 years of age.
Despite its clinical significance, Ewing sarcoma remains a rare entity. The overall incidence is less than 1 per 1 million population per year, accounting for approximately 9% of all primary malignancies of bone. While the disease has been reported across a vast age spectrum—ranging from infancy to the elderly—the overwhelming majority of cases manifest in patients between 5 and 25 years of age.
Anatomic Distribution and Demographics
The anatomic predilection of Ewing sarcoma heavily favors the appendicular and axial skeleton. The most common locations include:
* Long Bones: Typically originating in the metaphyses with frequent, extensive extension into the diaphysis.
* Flat Bones: High incidence in the pelvic girdle (ilium, ischium, pubis) and the shoulder girdle (scapula).
* Atypical Locations: Rarely, the tumor may present in the vertebral column or the small bones of the hands and feet.
Demographically, Ewing sarcoma exhibits a slight male predominance, a characteristic shared with most other bone sarcomas. Notably, there is a striking racial disparity: Ewing sarcoma is exceedingly rare in individuals of African descent. Currently, there are no known environmental or genetic predisposing factors, distinguishing it from conditions like osteosarcoma (which may be linked to retinoblastoma or Li-Fraumeni syndrome).
Clinical Presentation and Diagnostic Challenges
The clinical presentation of Ewing sarcoma is notoriously insidious, frequently leading to significant delays in diagnosis. Pain is an almost universal chief complaint. Initially, this pain may be mild, intermittent, and responsive to conservative measures such as nonsteroidal anti-inflammatory drugs (NSAIDs) or rest.
Clinical Pitfall: The average delay from the onset of symptoms to definitive diagnosis is a staggering 34 weeks. Studies indicate an average patient-dependent delay of 15 weeks before seeking medical attention, compounded by an average physician-dependent delay of 19 weeks from the initial consultation to the correct diagnosis.
This profound delay underscores the absolute necessity of obtaining high-quality orthogonal radiographs at the initial visit for any child or young adult presenting with persistent bone pain, and critically, re-evaluating with repeat imaging if symptoms fail to resolve.
The Osteomyelitis Mimic
Ewing sarcoma frequently masquerades as an infectious process. Patients may present with systemic and localized signs suggesting acute or subacute osteomyelitis, including:
* Low-grade fever
* Localized erythema and warmth
* Palpable swelling and tenderness
* Elevated inflammatory markers: Increased white blood cell (WBC) count, elevated erythrocyte sedimentation rate (ESR), and elevated C-reactive protein (CRP).
Surgical Warning: A needle aspirate of a Ewing sarcoma may yield necrotic, liquefied tumor tissue that grossly resembles purulent exudate. If this material is mistakenly assumed to be an abscess, the entire specimen might be sent exclusively to microbiology. Rule of Thumb: All biopsy and aspirate specimens obtained from suspected infectious or neoplastic lesions must be sent for both microbiological culture and rigorous histopathological analysis.
Diagnostic Imaging and Staging
Accurate staging is the cornerstone of formulating a multidisciplinary treatment plan. Ewing sarcoma requires a comprehensive, systemic imaging approach.
Plain Radiography
Classically, Ewing sarcoma presents radiographically as a destructive, permeative, or moth-eaten radiolucent lesion in the diaphysis of a long bone, accompanied by a lamellated or "onion skin" periosteal reaction. However, in clinical reality, the tumor more frequently originates in the metaphysis and extends a considerable distance into the diaphysis. In flat bones, such as the pelvis, the radiographic appearance is often that of a nonspecific, aggressive destructive lesion.
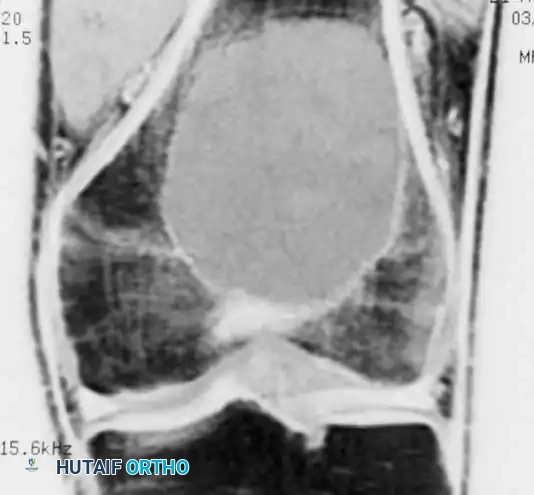
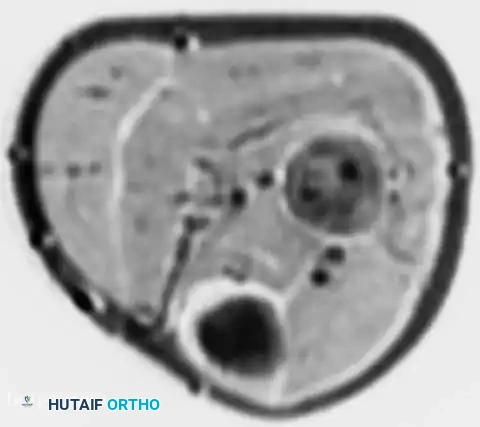
Fig. 22-9 A and B, Anteroposterior and lateral radiographs of the left fibula of a 7-year-old girl with Ewing sarcoma. Involvement of a large portion of the bone (or even the entire bone) is typical of Ewing sarcoma. C, MRI shows a large soft-tissue mass. D, Typical microscopic appearance of Ewing sarcoma.
Advanced Cross-Sectional Imaging
- Magnetic Resonance Imaging (MRI): An MRI of the entire involved bone is mandatory. While "skip" metastases (common in osteosarcoma) are not a recognized feature of Ewing sarcoma, it is highly common for a massive portion of the marrow cavity—or the entire bone—to be involved. MRI is unparalleled in defining the intramedullary extent and delineating the extraosseous soft-tissue mass, which is often disproportionately large compared to the degree of cortical destruction.
- Computed Tomography (CT): A baseline high-resolution CT scan of the chest is critical, as the lungs are the most common site of distant metastasis.
- Bone Scintigraphy: A Technetium-99m whole-body bone scan is required to evaluate for skeletal metastases, which represent the second most common site of systemic spread.
- Bone Marrow Aspiration and Biopsy: In stark contrast to the staging protocols for most other primary bone sarcomas, bilateral bone marrow aspirates and biopsies are a routine and mandatory component of Ewing sarcoma staging to definitively rule out diffuse systemic marrow involvement.
Pathology and Molecular Genetics
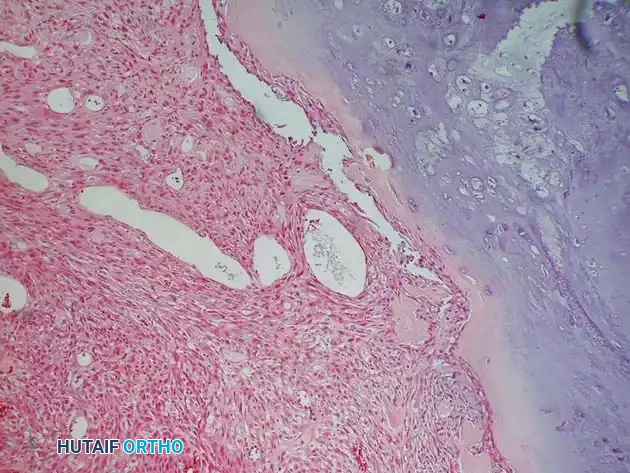
Histologically, Ewing sarcoma is the quintessential "small round blue cell tumor." The microscopic architecture consists of dense sheets of uniform, small, round cells with scant cytoplasm and very little intercellular matrix.
Because the light microscopic appearance is highly nonspecific, advanced cytogenetic and immunohistochemical (IHC) studies are absolute requirements to differentiate Ewing sarcoma from other small blue cell tumors of childhood (e.g., lymphoma, neuroblastoma, embryonal rhabdomyosarcoma).
Molecular Translocations
Ewing sarcoma is defined by specific chromosomal translocations that fuse the EWSR1 gene on chromosome 22 with various members of the ETS family of transcription factors.
* t(11;22)(q24;q12): The most common translocation, resulting in the EWS-FLI1 fusion protein, present in >90% of cases.
* Alternative Translocations: t(21;22)(q22;q12) [EWS-ERG] and t(7;22)(p22;q12) [EWS-ETV1] are also diagnostic.
Immunohistochemistry and Staining
- Positive Markers: Ewing sarcoma cells strongly express the MIC2 gene product (CD99) in a distinct membranous pattern. The cells are also typically Periodic Acid–Schiff (PAS) positive due to abundant intracellular glycogen.
- Negative Markers: The tumor is reticulin negative.
Differential Diagnosis via IHC
- Lymphoma: PAS negative, reticulin positive. Stains positive for Leukocyte Common Antigen (LCA/CD45) and specific T/B cell antigens.
- Embryonal Rhabdomyosarcoma: Stains positive for desmin, myoglobin, and muscle-specific actins.
- Hemangiopericytoma: Stains positive for Factor VIII.
- Metastatic Carcinoma/Melanoma: Stains positive for cytokeratins or melanocytic markers (HMB-45, Melan-A).
Prognostic Factors
The prognosis of Ewing sarcoma is dictated by a combination of clinical, laboratory, and histological parameters.
- Distant Metastases: The presence of distant metastasis at presentation is the single worst prognostic factor. Even with maximally aggressive systemic and local therapy, patients presenting with metastatic disease face a long-term survival rate of only 20%.
- Tumor Size and Location: Larger primary lesions correlate with poorer outcomes. Axial and proximal appendicular locations (e.g., pelvis, proximal femur) carry a worse prognosis than distal lesions, though this is heavily confounded by the fact that proximal tumors are typically much larger at the time of clinical detection.
- Age and Gender: Older age at presentation (typically defined as >12 to 15 years) and male gender are associated with inferior survival rates.
- Laboratory Values: Systemic symptoms (fever, anemia) and elevated baseline laboratory values (WBC, ESR, Lactate Dehydrogenase [LDH]) indicate high tumor burden and portend a worse prognosis.
- Histological Response to Chemotherapy: Similar to osteosarcoma, the degree of tumor necrosis following neoadjuvant chemotherapy is a profound predictor of overall survival.
- Genetics: While the specific translocation type (e.g., t(11;22) vs. t(21;22)) does not significantly alter the clinical course, secondary genetic alterations, such as aberrant p53 expression or STAG2 mutations, are emerging as important indicators of aggressive disease.
Note: Histological grade is not a prognostic variable, as all Ewing sarcomas are universally classified as high-grade malignancies.
Multidisciplinary Treatment Protocols
The management of Ewing sarcoma is strictly multidisciplinary. Because Ewing sarcoma is considered a systemic disease from inception (with presumed micrometastases), treatment must include systemic chemotherapy.
Systemic Chemotherapy
Before the advent of multiagent chemotherapy, the long-term survival rate for Ewing sarcoma treated with local therapy alone was a dismal <10%. Today, utilizing alternating cycles of VDC (Vincristine, Doxorubicin, Cyclophosphamide) and IE (Ifosfamide, Etoposide), modern centers report long-term survival rates of 60% to 75% for localized disease.
Local Control: Surgery vs. Radiation
Ewing sarcoma is highly radiosensitive, making radiotherapy a viable option for local control. However, the choice between surgical resection and definitive radiation is highly nuanced and must be individualized.
- Surgical Resection: Wide surgical resection is generally preferred if it can be achieved with negative margins and an acceptable functional outcome. Surgery avoids the long-term risks of high-dose radiation, including secondary radiation-induced sarcomas, joint contractures, and severe growth plate arrest in young children. Reports indicate a local recurrence rate of <10% following wide resection.
- Radiotherapy: Radiation is reserved for tumors in anatomically complex locations where wide surgical margins are impossible without unacceptable morbidity (e.g., massive pelvic or spinal tumors). It is also utilized as an adjuvant therapy if surgical margins are marginal or contaminated.
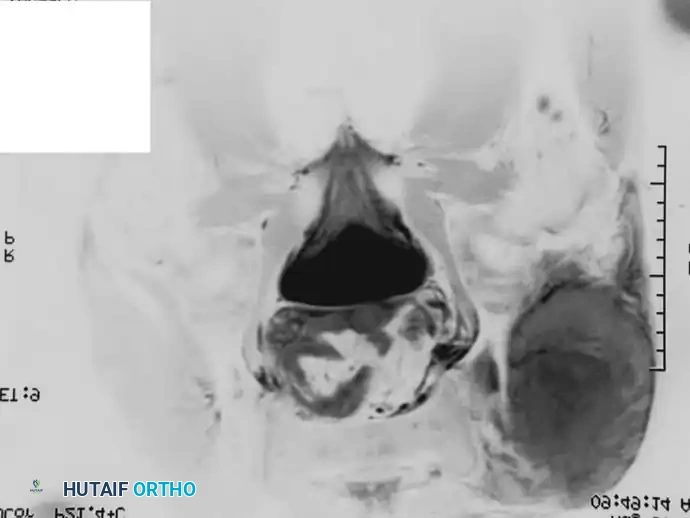
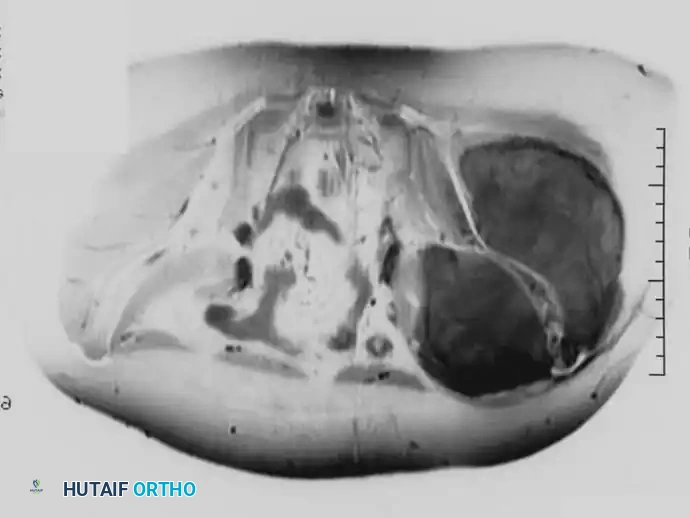
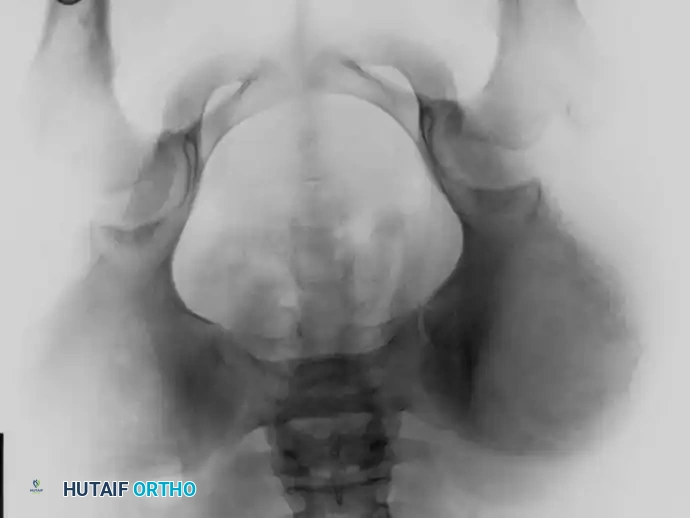
Fig. 22-10 A, Anteroposterior radiograph of a 13-year-old girl with Ewing sarcoma of the left hemipelvis. B, Bone scan. C and D, Axial and coronal MRI show the full extent of the lesion. Because of the severe morbidity associated with the surgical management of a large tumor in this location, the patient was treated with chemotherapy and definitive radiation; 3.5 years after treatment, there was no evidence of disease.
Surgical Management: Step-by-Step Principles
Preoperative Restaging
Following the completion of neoadjuvant chemotherapy, repeat staging studies (MRI of the primary site, CT chest) are mandatory.
* Radiographic Response: Repeat plain films often demonstrate increased ossification and sclerosis of the lesion, indicating a positive treatment response.
* MRI Response: A marked reduction in the extraosseous soft-tissue mass is typically observed, which may convert an initially unresectable tumor into a surgical candidate.
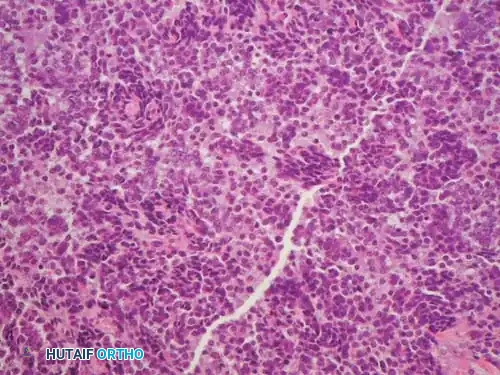
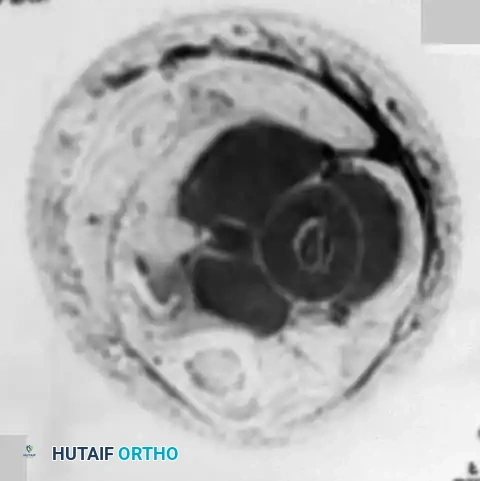
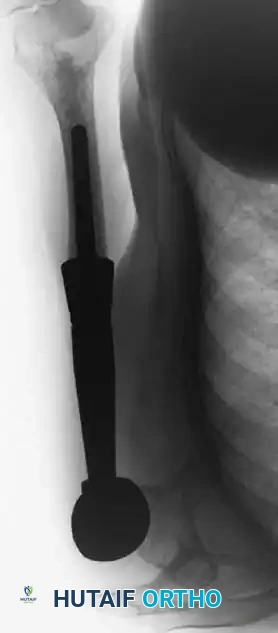
Fig. 22-9 (Continued) E and F, Anteroposterior and lateral radiographs after completion of neoadjuvant chemotherapy. Note the increased ossification of the lesion. G, Repeat MRI after neoadjuvant chemotherapy shows a marked reduction in the size of the soft-tissue mass. H and I, Anteroposterior and lateral radiographs of the left tibia after wide resection of the tumor. The distal fibular physis was preserved. Wide resection avoids complications associated with radiation in a growing child.
Surgical Approach and Resection
- Biopsy Tract Excision: The previous biopsy tract must be elliptically excised en bloc with the definitive tumor specimen to prevent local seeding.
- Exposure and Neurovascular Protection: The surgical approach is dictated by the anatomic compartment. The primary goal is to identify and protect major neurovascular bundles early in the dissection. If the neurovascular bundle is encased by the tumor and cannot be freed with a normal tissue cuff, amputation may be required.
- Wide Margins: The tumor must be resected with a continuous cuff of normal, healthy tissue in all dimensions (wide margin). Intralesional or marginal resections are oncologically unacceptable and carry a high risk of local recurrence.
- Osteotomy: Bone cuts are planned based on the preoperative MRI, typically aiming for a 3 to 5 cm margin beyond the intramedullary extent of the tumor.
Reconstruction
Reconstruction depends on the patient's age, skeletal maturity, and the anatomic site:
* Endoprosthetics: Modular megaprostheses are commonly used for metaphyseal/articular resections in older adolescents and adults. Expandable prostheses are utilized in skeletally immature patients to accommodate future growth.
* Biological Reconstruction: Intercalary allografts, vascularized fibular autografts, or allograft-prosthetic composites may be used for diaphyseal defects.
Relapse and Survival
Disease relapse carries a grim prognosis. Patients experiencing a local recurrence have an approximate 5-year survival rate of 20%, whereas those relapsing with distant metastases face a 5-year survival rate of roughly 10%. Time to relapse is critical; relapse within the first year after primary treatment indicates highly chemoresistant disease and a profoundly poor prognosis compared to late relapses.
Differential Considerations and Secondary Sarcomas: The Case of Chondrosarcoma
While Ewing sarcoma dominates the differential for young patients, older adults presenting with destructive bone lesions require a different diagnostic paradigm. It is crucial to distinguish primary high-grade sarcomas from malignant degeneration of benign lesions or dedifferentiated variants of other sarcomas, such as chondrosarcoma.
Dedifferentiated Chondrosarcoma
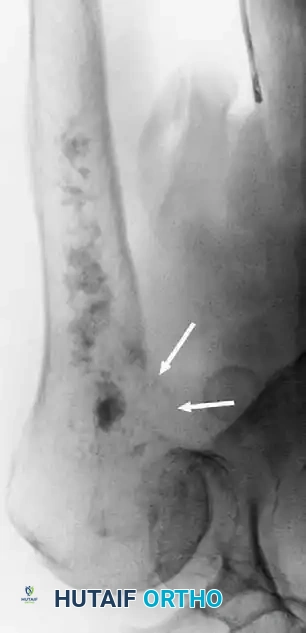
Chondrosarcoma is generally a disease of adulthood. A critical variant is dedifferentiated chondrosarcoma, characterized by a high-grade spindle cell sarcoma (often resembling osteosarcoma, fibrosarcoma, or undifferentiated pleomorphic sarcoma) arising directly adjacent to a pre-existing low-grade cartilaginous tumor.
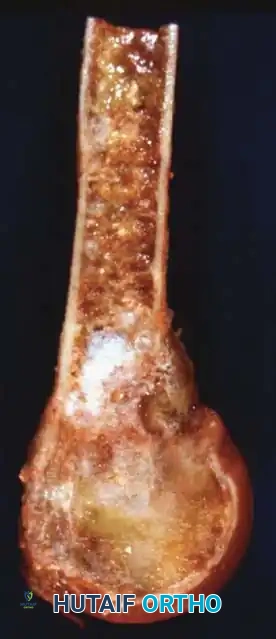
Fig. 22-8 A, Anteroposterior view of the right proximal humerus of a 92-year-old woman with dedifferentiated chondrosarcoma shows an aggressive-appearing area (arrows) adjacent to an otherwise typical chondrosarcoma. B, Resected specimen shows these features. C, Typical microscopic appearance of dedifferentiated chondrosarcoma. High-grade spindle cell sarcoma is located adjacent to low-grade chondrosarcoma. D, Anteroposterior radiograph after reconstruction with an endoprosthesis.
Surgical Principles in Chondrosarcoma
Unlike Ewing sarcoma, conventional chondrosarcoma is notoriously resistant to both chemotherapy and radiotherapy.
* Surgical Resection: The mainstay of treatment is wide surgical resection. Cartilage tumors transplant easily into soft tissues; therefore, the local recurrence rate following intraoperative tumor contamination is exceptionally high.
* Biopsy Considerations: For lesions in an expendable location (e.g., rib, fibula, clavicle), primary wide resection without a prior biopsy may be indicated to entirely eliminate the risk of tumor tract contamination.
* Prognosis: Following uncontaminated wide resection, the local recurrence rate is less than 10%. Prognosis depends heavily on histological grade and surgical margins. Patients with low-grade lesions enjoy a >90% 10-year survival rate. Conversely, high-grade conventional chondrosarcomas carry a 20% to 40% 10-year survival rate.
* Dedifferentiated Variant: The prognosis for dedifferentiated chondrosarcoma is catastrophic, with a 5-year survival rate of less than 15%, and most mortalities occurring within the first 24 months due to rapid pulmonary metastasis. Because low-grade chondrosarcomas are slow-growing, local recurrences and pulmonary metastases may not become clinically apparent until many years post-resection, necessitating lifelong surveillance.
Conclusion
The management of Ewing sarcoma requires a highly orchestrated, multidisciplinary approach. Early recognition of its insidious presentation, rigorous staging, and the integration of systemic neoadjuvant chemotherapy with precise, margin-negative surgical resection or definitive radiotherapy are paramount. As molecular diagnostics continue to evolve, targeted therapies addressing specific genetic translocations hold the promise of further improving survival rates for this aggressive malignancy.
📚 Medical References
- Ewing sarcoma in children and adolescents, J Bone Joint Surg 77A:54, 1995.
- Asavamongkolkul A, Eckardt JJ, Eilber FR, et al: Endoprosthetic reconstruction for malignant upper extremity tumors, Clin Orthop Relat Res 360:207, 1999.
- Augereau B, Wioland M, DeLabriolle-Vaylet C, et al: The intraoperative radioisotopic localization of
You Might Also Like