Surgical Management of Giant Cell Tumors and Benign Fibrous Hand Lesions
Key Takeaway
Giant cell tumors of the tendon sheath (GCTTS) are the most common solid cellular tumors of the hand. Despite their benign nature, they exhibit a high recurrence rate of up to 27% due to complex anatomical involvement around neurovascular bundles and flexor tendons. This guide details the pathological characteristics, surgical indications, and meticulous microsurgical techniques required for complete extirpation and minimization of recurrence in GCTTS and other benign fibrous hand lesions.
GIANT CELL TUMOR OF THE TENDON SHEATH (XANTHOMA)
Giant cell tumors of the tendon sheath (GCTTS), historically referred to as xanthomas, were first comprehensively described in the surgical literature by Beekman in 1915. Today, they are universally recognized by orthopedic oncologists and hand surgeons as the most common solid cellular tumors of the hand, second only to simple ganglion cysts in overall prevalence among hand masses.
These lesions exhibit a broad demographic distribution, presenting in patients ranging from 8 to 80 years of age, with a peak incidence in the third to fifth decades of life. GCTTS demonstrates a distinct anatomical predilection for the hand compared to any other anatomical region. Topographically, they most frequently manifest as firm, lobulated, and non-transilluminating masses on the volar-lateral aspect of the index and middle fingers. It is also noteworthy that the presence of multiple xanthomas may serve as a clinical indicator of underlying familial hypercholesterolemia, necessitating appropriate metabolic workup.
Clinical Presentation and Pathophysiology
The natural history of a giant cell tumor of the tendon sheath is characterized by an indolent, slow-growing trajectory. These masses can remain static in size for years. Pain and localized tenderness are exceptionally rare unless the tumor undergoes rapid expansion or mechanically impinges upon adjacent neurovascular structures.
When GCTTS arises in close proximity to an articulation—most commonly the proximal interphalangeal (PIP) joint or distal interphalangeal (DIP) joint—it can achieve sufficient volume to mechanically block joint excursion, leading to a progressive loss of range of motion. In advanced or neglected cases, the tumor may exert pressure on the underlying cortex, resulting in pressure erosions of the bone, a finding visible on standard radiographs.
Clinical Pearl: Radiographic bone erosion in GCTTS is typically a smooth, scalloped, extrinsic pressure erosion with a sclerotic margin, distinguishing it from the aggressive, permeative intrinsic bone destruction seen in malignant osseous neoplasms.
Grossly, the tumor presents as a well-encapsulated, lobular mass with a characteristic yellow, tan, or mottled brown appearance, dictated by its lipid and hemosiderin content. Histological examination is definitive, revealing a complex admixture of:
* Proliferating mononuclear spindle cells.
* Dense fibrous stroma.
* Cholesterol-laden histiocytes (xanthoma cells).
* Multinucleated giant cells (morphologically resembling osteoclasts).
* Intracellular and extracellular hemosiderin deposits.
Surgical Management and Recurrence Mitigation
Although histologically benign, GCTTS is notorious for its high local recurrence rate, historically reported at approximately 27%, even following what appears to be a meticulous gross total excision. The friable nature of the tumor fragments makes microscopic residual disease a constant intraoperative threat.
Risk Factors for Recurrence:
1. Presence of adjacent degenerative joint disease (DJD).
2. Location at the distal interphalangeal (DIP) joint of the fingers or the interphalangeal (IP) joint of the thumb.
3. Radiographic evidence of bony invasion or cortical erosion.
4. Multifocal or satellite disease.
5. Inadequate surgical technique (e.g., marginal excision without magnification).
Surgical Technique:
Excision is frequently highly complex. The tumor lobules have a propensity to wind intimately around and between the flexor tendons, infiltrate the synovial sheaths, encircle the digital nerves and arteries, and occasionally extend dorsally to involve the extensor apparatus. In severe cases, the tumor may encompass three-fourths of the digit's circumference.
- Anesthesia and Positioning: The procedure must be performed under regional or general anesthesia. A well-padded pneumatic tourniquet is absolute mandatory to provide a bloodless surgical field, allowing for precise identification of microvascular structures.
- Incision and Exposure: Extensile surgical approaches are required. A Brunner zigzag incision or a mid-lateral incision is preferred to allow full exposure of the flexor tendon sheath and neurovascular bundles without crossing flexion creases at a right angle.
- Dissection: Gentle, blunt dissection is paramount to minimize the fragmentation of the encapsulated tumor mass. The digital nerve and artery must be identified proximally in normal tissue and traced distally through the tumor bed.
- Magnification: The use of surgical loupes (minimum 3.5x to 4.5x magnification) or an operating microscope is strongly recommended. Magnified vision is critical to identify and resect discolored, satellite synovial tumor deposits, which must be excised en bloc with the primary specimen to prevent recurrence.
Despite these challenges, GCTTS is considered a surgically curable lesion provided that complete, unfragmented extirpation is achieved.
BENIGN TUMORS OF FIBROUS ORIGIN
Fibrous tissue proliferation in the hand is frequently a physiological response to local trauma, often resulting in simple scar tissue. Consequently, the definitive diagnosis of a true fibrous tumor relies heavily on a synthesis of the tumor's gross appearance, the patient's age, the lesion's clinical behavior, and precise histological evaluation.
While all tumors of fibrous origin can manifest in the hand, and the vast majority are benign, they frequently exhibit active or locally aggressive biological behavior, carrying a significant tendency to recur following local excision. The differential diagnosis for benign fibrous tumors of the hand is extensive and includes:
* Simple Fibroma
* Recurring Digital Fibrous Tumor of Childhood (Infantile Digital Fibromatosis)
* Juvenile Aponeurotic Fibroma
* Dupuytren's contractures or nodules
* Fibromatosis (Extra-abdominal desmoid tumors)
* Pseudosarcomatous fasciitis
Surgical Warning: Fibromatosis (extra-abdominal desmoid) and pseudosarcomatous fasciitis are highly aggressive local lesions. Unlike simple fibromas, they typically involve the more proximal portions of the upper extremity and require wide oncologic resection margins to prevent devastating local recurrence.
Intraneural Lipofibromas
Intraneural lipofibromas (also known as fibrolipomatous hamartomas) are exceedingly rare, benign tumors that exhibit a striking predilection for the median nerve. Patients typically present within the first three decades of life. The classic clinical presentation involves a slowly enlarging, often doughy mass located in the palmar aspect of the hand or the volar wrist.
As the fibro-fatty tissue proliferates within the nerve sheath, patients may develop symptoms of altered median nerve function, mimicking severe carpal tunnel syndrome, including sensory deficits and thenar motor weakness.
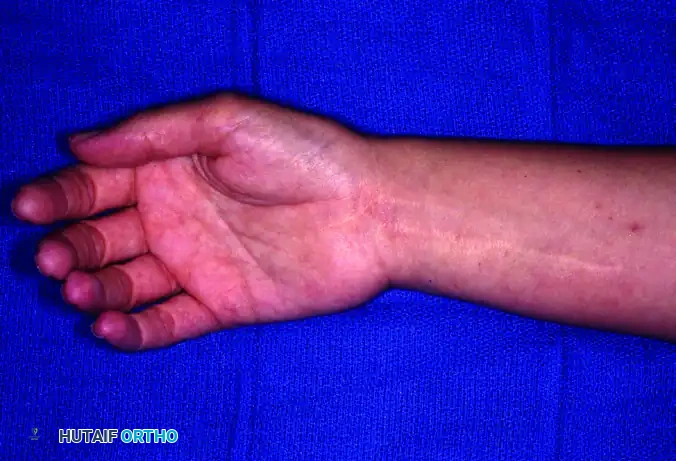
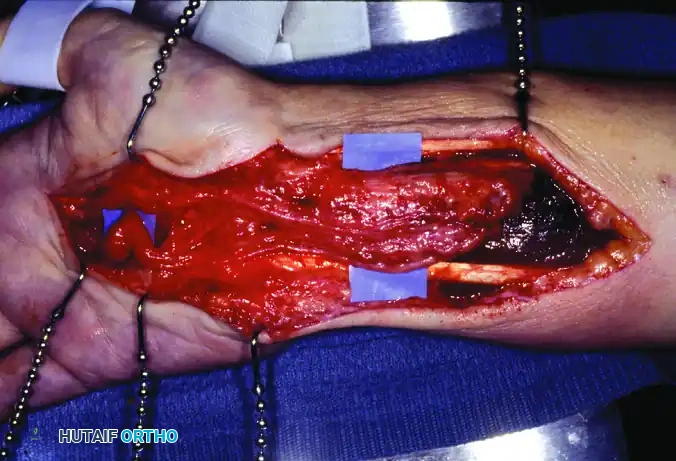
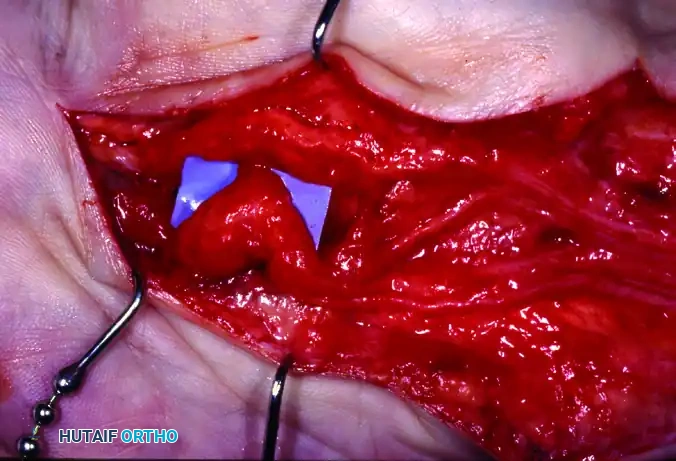
Figure 77-2: (A) Clinical presentation of an intraneural lipofibroma of the median nerve, demonstrating characteristic palmar and distal forearm enlargement. (B) Intraoperative surgical exposure revealing extensive lipofibromatous changes infiltrating the main trunk and common digital nerves following decompression and external neurolysis of the median nerve. (C) High-magnification view detailing the gross enlargement and fibro-fatty infiltration of the digital nerve branches.
Microscopically, the pathology is defined by the infiltration of mature fibroadipose tissue into the epineurium and perineurium, separating and compressing the individual nerve fascicles.
Surgical Management of Lipofibromas:
The surgical approach to intraneural lipofibromas is highly controversial and fraught with risk. Because the adipose tissue is intimately interspersed between the nerve fascicles, attempting to dissect the tumor out of the nerve (intraneural dissection) inevitably leads to catastrophic ischemic injury and permanent loss of nerve function.
Initial management should consist of prophylactic decompression (e.g., extended carpal tunnel release) and external neurolysis. However, in cases of intractable pain or complete loss of function, en bloc resection of the involved nerve segment followed by nerve grafting may be the only viable salvage procedure.
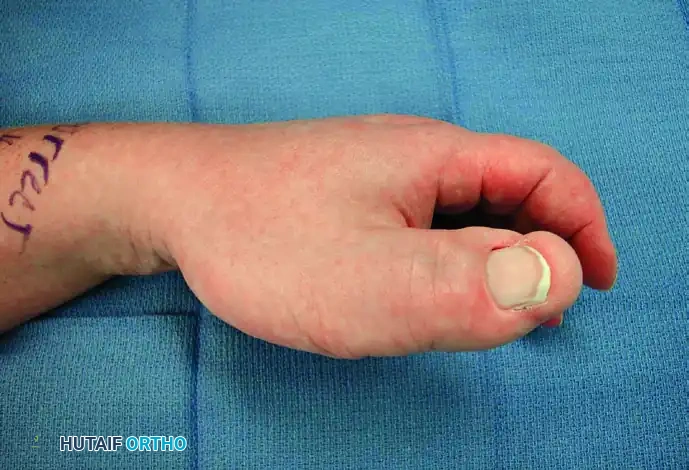
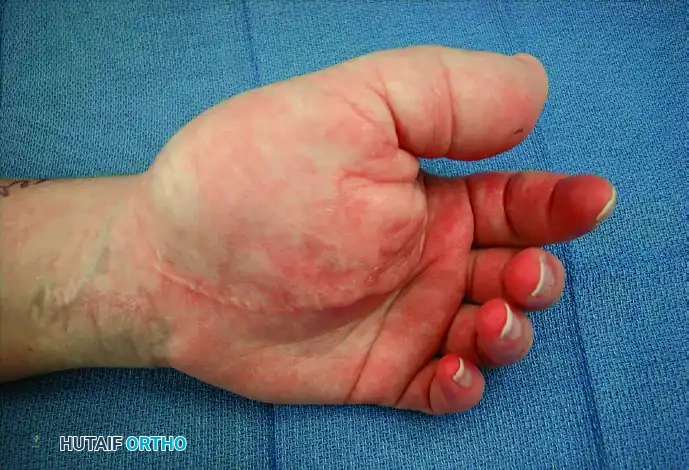
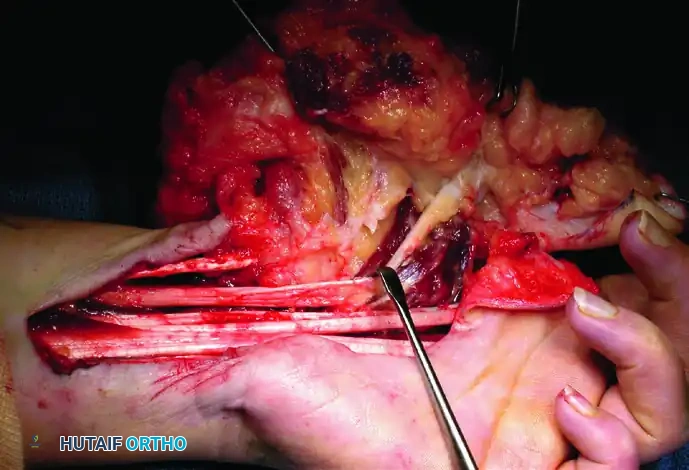
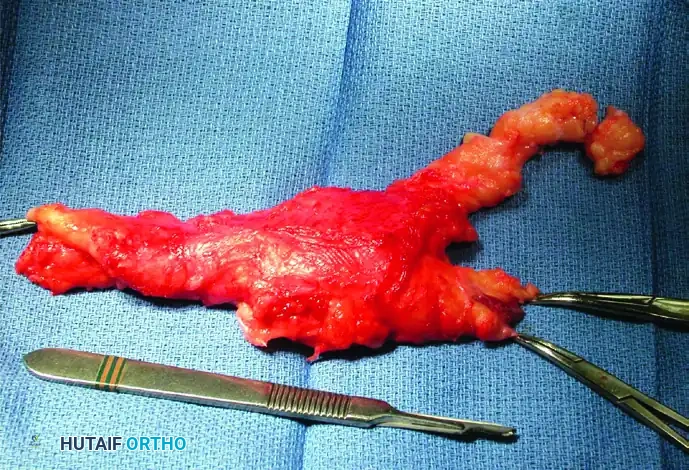
Figure 77-3: (A and B) Clinical views of a 25-year-old woman presenting with intractable neuropathic pain following multiple failed debulking procedures for a median nerve lipofibroma. (C) Complex intraoperative nerve dissection in the distal forearm and hand, highlighting the extensive neural involvement. (D) The gross resected specimen. Note: Pain relief is rarely complete with debulking alone; complete nerve excision and grafting should be considered the final step in management when function is lost or pain is debilitating, barring malignant degeneration.
While the median nerve is the most common site, these lesions can occasionally affect other nerves, such as the ulnar digital nerves.
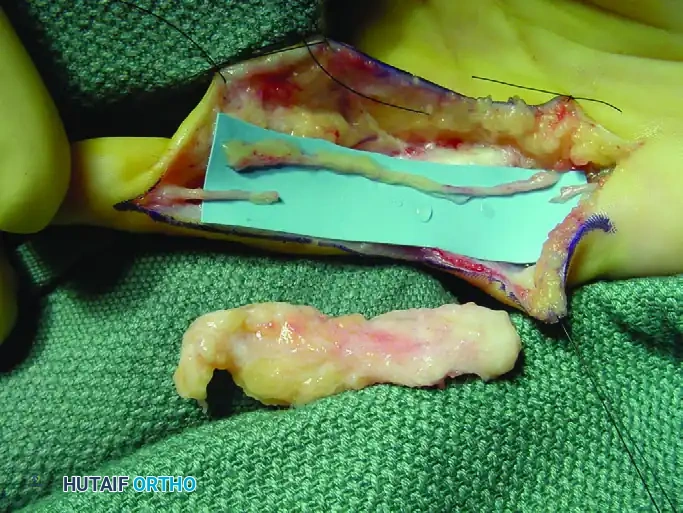
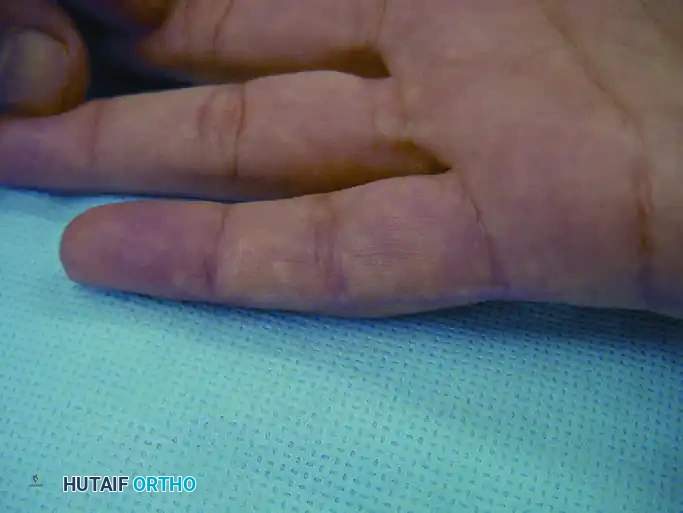
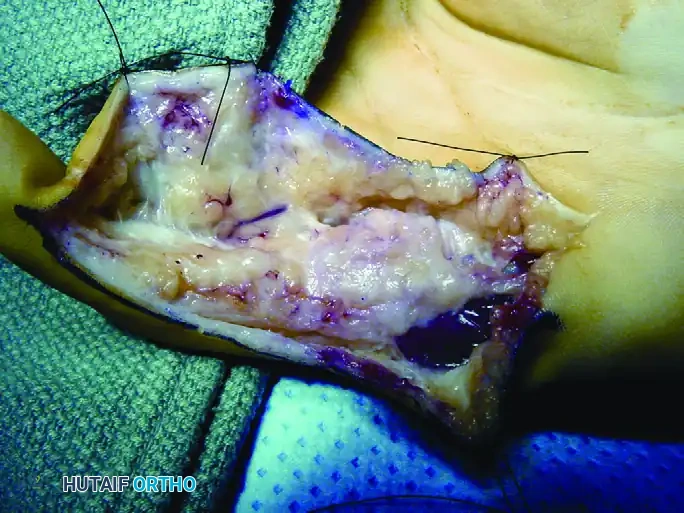
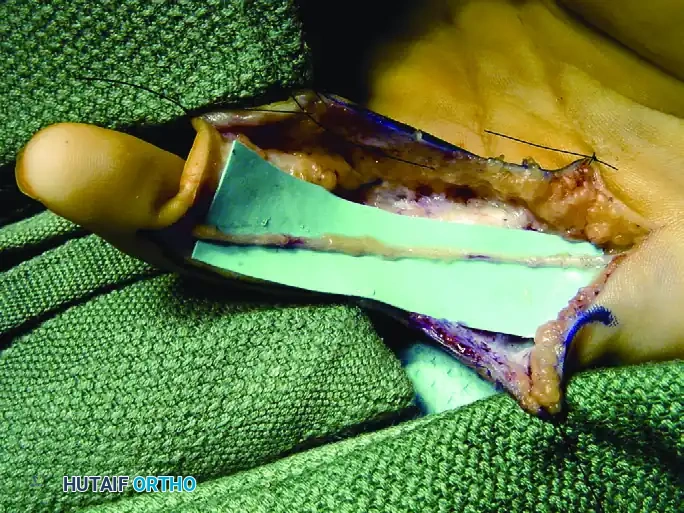
Figure 77-4: (A) Clinical presentation of a recurrent intraneural lipofibroma affecting the ulnar digital nerve to the small finger. (B) Surgical exposure demonstrating a 4-cm segment of the involved ulnar digital nerve, flanked by normal-appearing proximal and distal nerve segments. (C) En bloc excision of the diseased digital nerve and tumor, with a harvested nerve graft placed in proximity for size matching. (D) The interpositional nerve graft meticulously sutured in place using microsurgical epineurial repair techniques.
Recurring Digital Fibrous Tumor of Childhood (Infantile Digital Fibromatosis)
First characterized by Reye in 1965, this benign fibrous tumor specifically develops in the fingers and toes of infants and young children. The pathognomonic histological feature of this lesion is the presence of intracytoplasmic inclusion bodies (composed of actin filaments) within the proliferating fibroblasts. Notably, these inclusion bodies are often invisible with routine hematoxylin and eosin (H&E) staining and require specialized stains (such as Masson trichrome or phosphotungstic acid-hematoxylin) or electron microscopy for definitive identification.
A viral etiology has been hypothesized due to the inclusion bodies, though this remains unproven. Clinically, these tumors exhibit a strong tendency to be multicentric, frequently occurring simultaneously on several digits. The primary site of involvement is the dermis, characteristically sparing the overlying epidermis.
Management:
Crucially, these lesions possess zero malignant potential, and spontaneous regression has been well-documented in the literature. Therefore, a conservative, observational approach is often the first line of management. Marginal excision is only recommended when the tumor's size severely compromises digital function, causes joint contractures, or results in severe cosmetic deformity (especially if tendons, joints, or the nail matrix are involved). Surgeons must counsel parents that local recurrence after marginal excision is exceptionally common, occurring in up to 60% of patients.
Juvenile Aponeurotic Fibroma (Calcifying Aponeurotic Fibroma)
Initially described by Keasbey in 1953, the juvenile aponeurotic fibroma is a benign, locally aggressive fibrous tumor that typically arises in the hands or wrists of children and young adults. It is frequently referred to as a "calcifying aponeurotic fibroma" because, as the patient ages, the cartilaginous component of the tumor undergoes stippled calcification—a distinct radiographic feature that helps differentiate it from other benign fibrous hand tumors.
Clinically, it presents as a painless, solitary, and somewhat mobile mass, usually less than 4 cm in diameter. It demonstrates a strong predilection for the volar aspect (palm) of the hand and is intimately connected to peritendinous tissues and the palmar fascia. Unlike Dupuytren's disease, it has no gender predilection and does not specifically favor the ulnar side of the hand.
Surgical Considerations:
Juvenile aponeurotic fibromas are notorious for their ability to infiltrate surrounding skeletal muscle and subcutaneous fat. Because this tumor has a distinct and high tendency for local recurrence following simple marginal excision—particularly in younger children where recurrence rates approach 50%—a wide local excision is the recommended surgical standard. However, this wide excision must be carefully balanced against the preservation of vital hand function; amputation or sacrifice of major motor units is rarely justified for this benign pathology.
Fibroma
True simple fibromas are relatively rare in the hand. They can manifest as deep lesions, often arising directly from a joint capsule, or as superficial subcutaneous masses. Demographically, they tend to occur early in life, exhibit a limited period of active growth, and then stabilize or subside. Unlike juvenile aponeurotic fibromas, simple fibromas do not exhibit calcifications on radiographs unless they have been present and subjected to microtrauma for an extended period.
Clinically, fibromas are easily distinguishable from Dupuytren's nodules:
* They present earlier in life.
* They do not tend to multiply or form cords.
* They lack a predilection for the ulnar digits.
* They are not associated with progressive flexion contractures of the digits.
Grossly, these tumors are firm, white, and composed of dense, mature fibroblasts interwoven with thick fibrous tissue. They are typically well-encapsulated. Surgical management consists of a marginal extracapsular excision, which is easily achieved via blunt dissection and is almost universally curative.
Neurofibroma
Neurofibromas in the hand rarely exist as isolated, solitary lesions. The vast majority of multiple lesions are syndromic, associated with Neurofibromatosis Type 1 (NF-1, von Recklinghausen disease). Solitary forms typically present in the first decade of life, whereas the multiple syndromic forms frequently become clinically apparent or symptomatic after 30 years of age.
In the hand, these lesions frequently involve the more distal digital nerves. The progressive enlargement of the nerve can produce grotesque angular deformities of the fingers and localized macrodactyly (gigantism). Clinically, neurofibromas are centrally located along the nerve axis, non-tender, nodular, and lack a true capsule. They frequently infiltrate the overlying skin, making them significantly less mobile than schwannomas (neurilemomas).
Pitfall: Unlike a schwannoma, which grows eccentrically and can often be enucleated while preserving the main nerve fascicles, a neurofibroma is a plexiform mass of irregular, thickened nerve fibers separated by an abundant endoneural matrix. It is intimately intertwined with the functional nerve fibers.
Surgical Excision:
Because the nerve fibers are interspersed directly within the tumor mass, a neurofibroma is generally not resectable without sacrificing the involved nerve elements. Often, these tumors involve cutaneous sensory branches where the nerve caliber appears completely normal immediately proximal and distal to the lesion.
If the lesion exceeds 2.0 cm, or if there is rapid growth or onset of rest pain, a biopsy is mandatory to rule out malignant peripheral nerve sheath tumor (MPNST) degeneration, which carries a metastatic rate exceeding 25%. For benign, symptomatic lesions, meticulous and complete en bloc excision of the involved nerve segment is required. While complete excision is curative, reoperation rates of 12% to 24% have been reported, almost exclusively resulting from incomplete initial tumor extirpation. Reconstruction with interpositional nerve grafting should be performed concurrently if a critical sensory or motor territory is sacrificed.
📚 Medical References
You Might Also Like
