Comprehensive Surgical Management of Benign Cartilaginous Bone Tumors: Chondromyxoid Fibroma and Chondroblastoma
Key Takeaway
Chondromyxoid fibroma is a rare benign cartilaginous bone tumor, representing less than 0.5% of all bone neoplasms. Typically presenting in the proximal tibia of patients aged 10 to 30, it requires meticulous surgical management. This guide details the evidence-based approach to diagnosis, radiographic evaluation, and surgical treatment via extended curettage and bone grafting, alongside related cartilaginous lesions such as chondroblastoma.
INTRODUCTION TO BENIGN CARTILAGINOUS LESIONS
Benign tumors of cartilaginous origin present a unique diagnostic and therapeutic challenge in orthopedic oncology. Among these, Chondromyxoid Fibroma (CMF) and Chondroblastoma (CB) are distinct entities that, while histologically benign, exhibit locally aggressive behavior capable of causing significant structural compromise to the host bone.
Because these lesions frequently occur in the metaphyseal and epiphyseal regions of long bones in young, active patients, surgical management must balance the complete eradication of the tumor with the preservation of joint biomechanics and physeal integrity. This comprehensive guide delineates the clinical presentation, advanced radiographic evaluation, histopathological hallmarks, and step-by-step surgical management of these rare neoplasms, emphasizing the gold standard of extended intralesional curettage and defect reconstruction.
CHONDROMYXOID FIBROMA: CLINICAL AND PATHOLOGIC OVERVIEW
Epidemiology and Clinical Presentation
Chondromyxoid fibroma is an exceptionally rare lesion of cartilaginous origin. According to extensive institutional series, including data from the Mayo Clinic, CMF represents less than 0.5% of all primary bone tumors.
While the tumor may manifest at any age, the peak incidence occurs in the second and third decades of life (patients 10 to 30 years old). The proximal tibia is the most frequently involved anatomic site, though any bone may be affected.
Clinically, patients typically present with an insidious onset of localized, progressive pain. However, presentation varies by anatomic location. When the tumor arises in the small tubular bones of the hands or feet, the chief complaint is often a painless, palpable mass or localized swelling rather than mechanical pain.
Radiographic Evaluation
The radiographic signature of a chondromyxoid fibroma is that of a benign, yet locally expansile, neoplasm.
- Location: Lesions are classically eccentrically located in the metaphysis of long bones.
- Morphology: It presents as a well-circumscribed, radiolucent (lytic) lesion bordered by a distinct rim of reactive sclerosis.
- Internal Characteristics: The tumor frequently exhibits a trabeculated or "bubbly" appearance, which can closely mimic a nonossifying fibroma (NOF) or an aneurysmal bone cyst (ABC).
- Calcification: In stark contrast to other cartilaginous lesions (such as enchondromas or chondrosarcomas), radiographic evidence of intralesional calcification is typically absent in CMF. A rare exception is a surface (juxtacortical) variant of CMF, where abundant calcification may be observed.
Clinical Pearl: Because of its metaphyseal location and lytic, bubbly appearance, CMF must be included in the radiographic differential diagnosis of any such lesion, particularly when located in the proximal tibial metaphysis.
Histopathological Hallmarks
Accurate histopathological interpretation is paramount, as the cellular features of CMF can be easily misinterpreted by an inexperienced pathologist.
Microscopically, chondromyxoid fibroma exhibits a distinctly lobulated architecture. The center of these lobules is composed of loose, hypocellular myxoid tissue, while the periphery is characterized by hypercellular fibrous tissue. The background stroma often appears chondroid; however, distinct areas of mature hyaline cartilage are exceedingly rare. Microscopic, punctate calcification may occasionally be present despite its absence on plain radiographs.
Surgical Warning: CMF frequently contains areas with pleomorphic or atypical cells. It is critical that this cellular atypia does not lead to an erroneous, catastrophic misdiagnosis of chondrosarcoma, provided the lesion is otherwise radiographically and histologically consistent with CMF. Misdiagnosis can lead to unnecessary radical resection or amputation.
CHONDROBLASTOMA: THE EPIPHYSEAL COUNTERPART
While CMF is classically metaphyseal, Chondroblastoma is a related benign cartilaginous tumor that characteristically arises in the epiphysis or apophysis of long bones in skeletally immature patients. Because the surgical management (extended curettage) is virtually identical, it serves as a critical differential diagnosis.
Case 1: Proximal Humerus Chondroblastoma with Secondary ABC
A 16-year-old boy presented with a 1-year history of progressive left shoulder pain.
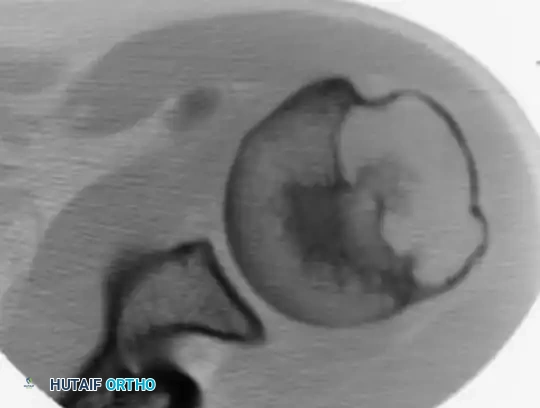
Fig. 21-4A: Anteroposterior radiograph of the left shoulder revealing a lytic lesion in the proximal left humerus extending across the open physis.
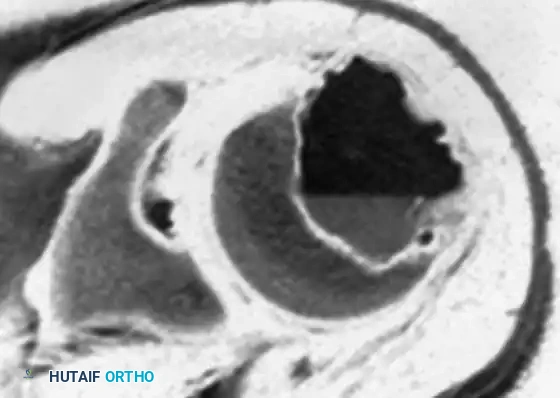
Fig. 21-4B: Axial CT scan demonstrating subtle internal calcification of the lesion, a hallmark of chondroblastoma ("chicken-wire" calcification).
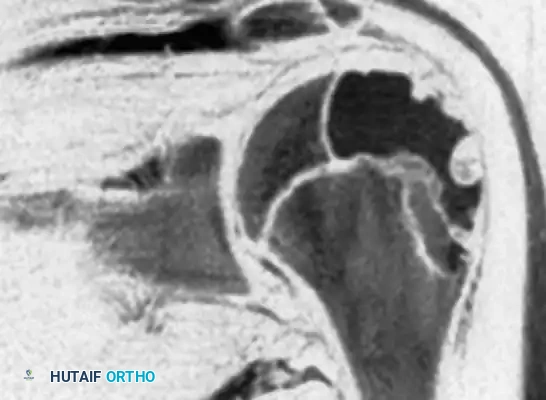
Fig. 21-4C: MRI showing the expansile nature of the lesion.
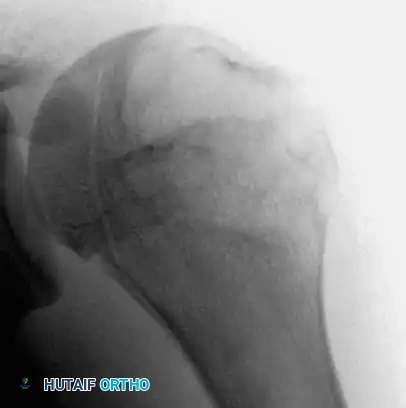
Fig. 21-4D: MRI demonstrating a distinct fluid-fluid level, indicative of a secondary aneurysmal bone cyst (ABC), which occurs in up to 20% of chondroblastomas.
An incisional biopsy confirmed the diagnosis of chondroblastoma with a secondary ABC. The patient underwent extended curettage and bone grafting.
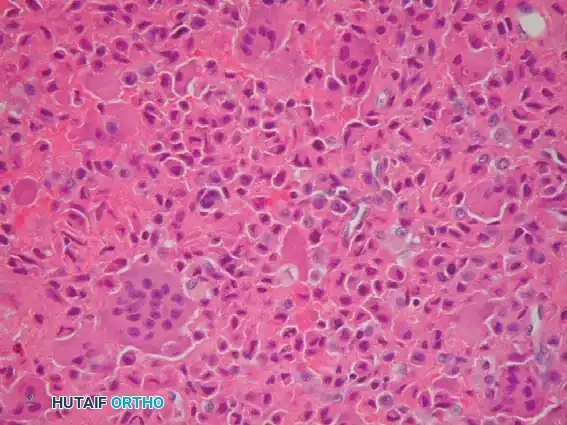
Fig. 21-4E: Postoperative anteroposterior radiograph demonstrating complete tumor eradication and successful incorporation of the bone graft.
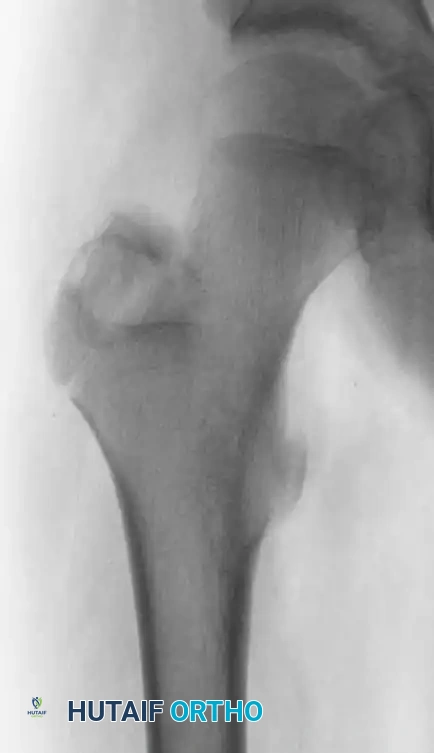
Fig. 21-4F: Typical microscopic appearance of chondroblastoma, characterized by mononuclear chondroblasts and scattered osteoclast-like giant cells.
Case 2: Greater Trochanter Chondroblastoma
A 12-year-old boy presented with worsening right hip pain persisting for several months. Apophyseal equivalents, such as the greater trochanter, are classic locations for chondroblastoma.
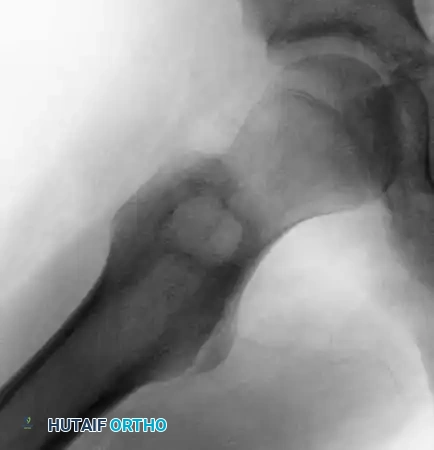
Fig. 21-5A: Anteroposterior radiograph of the right hip showing a radiolucent lesion in the greater trochanter.
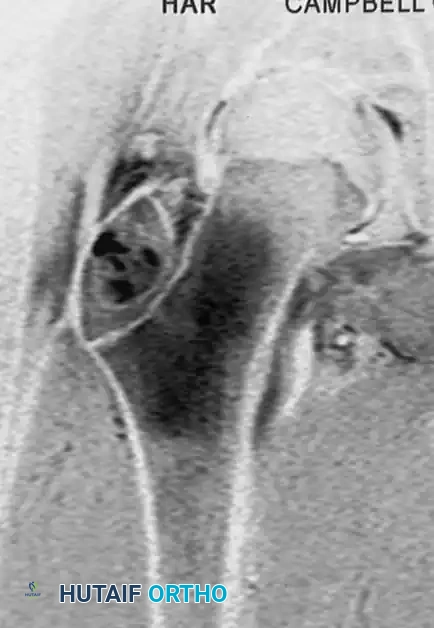
Fig. 21-5B: Lateral radiograph confirming the eccentric, lytic nature of the trochanteric lesion.
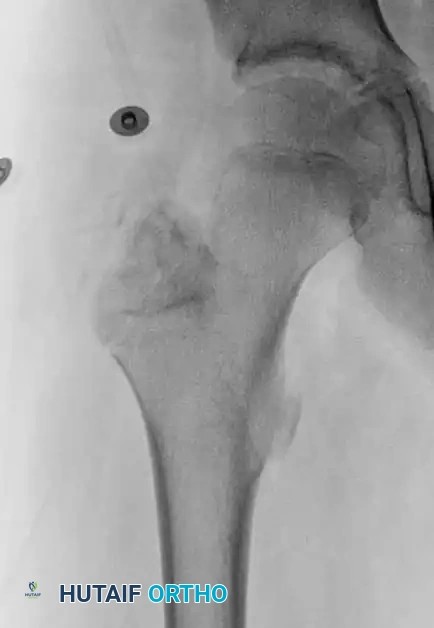
Fig. 21-5C: Coronal MRI revealing the lesion within the greater trochanter accompanied by extensive surrounding bone marrow edema, a classic MRI finding for chondroblastoma that correlates with the patient's pain.
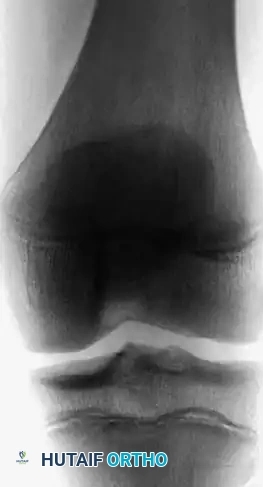
Fig. 21-5D: Postoperative radiograph following extended curettage and defect reconstruction using allograft cancellous bone chips combined with demineralized bone matrix (Allomatrix-C; Wright Medical Technology).
Case 3: Intercondylar Notch Chondroblastoma
A 14-year-old boy presented with worsening pain in the left knee, initially misdiagnosed as a meniscal pathology.
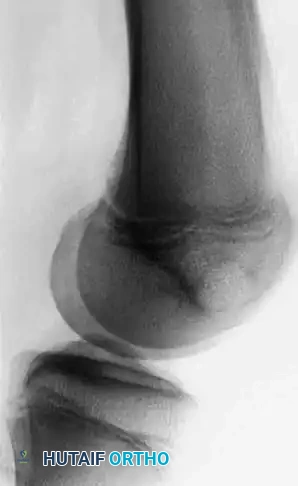
Fig. 21-6A: Anteroposterior radiograph showing a subtle lytic lesion in the distal femoral epiphysis.
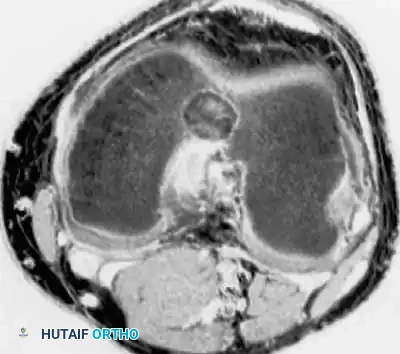
Fig. 21-6B: Lateral radiograph localizing the lytic lesion to the intercondylar notch.
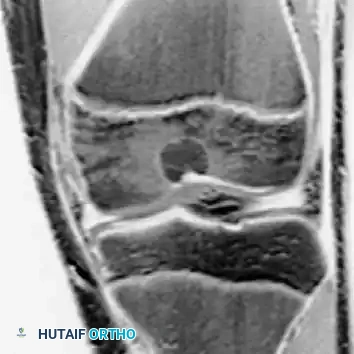
Fig. 21-6C: Axial MRI detailing the cortical breach and exact dimensions of the lesion.
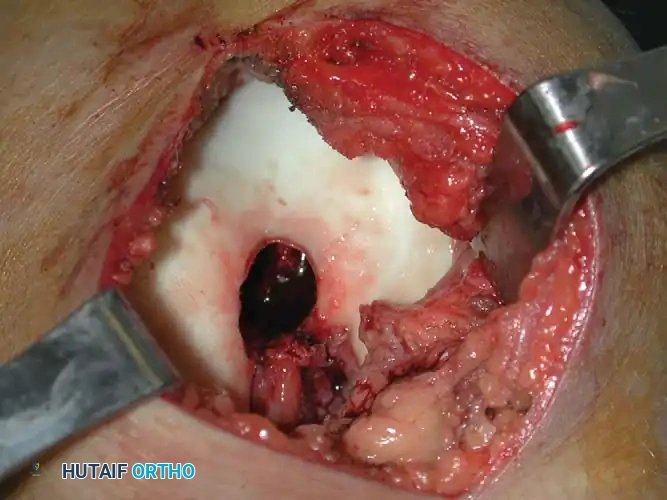
Fig. 21-6D: Coronal MRI showing the epiphyseal extent of the tumor, abutting the articular cartilage.
SURGICAL MANAGEMENT: EXTENDED CURETTAGE AND RECONSTRUCTION
The definitive treatment for both Chondromyxoid Fibroma and Chondroblastoma is surgical. Because these are benign but locally aggressive lesions, the goal is complete intralesional excision while preserving the structural integrity of the bone and adjacent joints.
Simple curettage is associated with unacceptably high recurrence rates. Therefore, extended curettage utilizing mechanical and chemical/thermal adjuvants is the gold standard. Local recurrence occurs in approximately 20% of CMF patients treated with curettage alone, necessitating meticulous technique. Sarcomatous change is exceedingly rare but has been reported, usually following prior radiation therapy.
1. Preoperative Planning and Positioning
- Imaging: High-quality MRI is mandatory to assess the exact tumor volume, cortical integrity, proximity to the physis (in skeletally immature patients), and subchondral bone stock.
- Biopsy: If the diagnosis is uncertain, a percutaneous core needle biopsy should be performed. The biopsy tract must be carefully planned so it can be excised during the definitive surgical approach.
- Positioning: The patient is positioned on a radiolucent table to allow for unhindered intraoperative fluoroscopy. A sterile tourniquet is applied for extremity lesions to ensure a bloodless surgical field, which is critical for visualizing residual tumor tissue.
2. Surgical Approach and Cortical Windowing
- An extensile approach is utilized based on the anatomic location.
- Once the bone is exposed, a large cortical window is created.
- Crucial Step: The cortical window must be at least as large as the maximum diameter of the underlying tumor. A "keyhole" approach is a common pitfall that inevitably leads to retained tumor in the peripheral recesses and subsequent local recurrence.
3. Intralesional Curettage
- The gross tumor is evacuated using a series of straight and angled curettes.
- Cartilaginous tumors often have a gritty, friable consistency. The surgeon must systematically curette the cavity in a sequential manner (e.g., clockwise) to ensure no quadrant is missed.
- Special attention must be paid to the subchondral bone plate. If the tumor abuts the joint, extreme care is taken not to breach the articular cartilage.
4. Mechanical and Thermal Adjuvants (Extended Curettage)
Once gross tumor is removed, the cavity undergoes extended curettage to eliminate microscopic disease residing in the cancellous bone trabeculae.
- High-Speed Burr: A high-speed motorized burr is used to aggressively abrade the walls of the cavity until normal, healthy bleeding cortical or cancellous bone is encountered.
- Thermal Coagulation: An Argon Beam Coagulator (ABC) or electrocautery is applied to the cavity walls. The argon beam provides a uniform depth of thermal necrosis (approximately 2-3 mm), effectively destroying residual microscopic tumor cells without compromising the structural integrity of the entire bone.
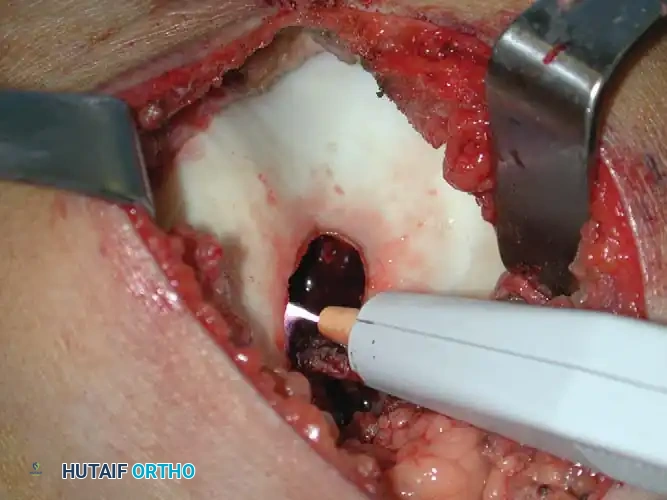
*Fig. 21-6E & F: (Left) Intraoperative photograph demonstrating the large cortical window and the cavity after gross curettage of the intercondylar notch lesion. (Right
📚 Medical References
- Adams M, Spencer G, Totterman S, et al: Case report 776, Skeletal Radiol 22:358, 1993.
- Baklouti S, Elleuch MH, Sellami F, et al: Chondromyxoid fi broma of the scapula: a case report, Int Orthop 16:193, 1992.
- Bullough PG: Bullough and Vigorita’s orthopaedic pathology, 3rd ed, London, 1997, Mosby-Wolfe. Cabral CEL, Romano S, Guedes P, et al: Chondromyxoid fi broma of the lumbar spine, Skeletal Radiol 26:488, 1997.
- Durr H, Lienemann A, Nerlich A, et al: Chondromyxoid fi broma of bone, Arch Orthop Trauma Surg 120:42, 2000.
- Forest M, Tomeno B, Vanel D, eds: Orthopedic surgical pathology: diagnosis of tumors and pseudotumoral lesions of bones and joints, Edinburgh, 1998, Churchill Livingstone. Fujiwara S, Nakamura I, Goto T, et al: Intracortical chondromyxoid fi broma of humerus: case report, Skeletal Radiol 32:156, 2003.
- Hau M, Fox E, Rosenberg A, et al: Chondromyxoid fi broma of the metacarpal: case report, Skeletal Radiol 30:719, 2001.
- Kenan S, Abdelwahab IF, Klein MJ, et al: Case report 837, Skeletal Radiol 23:237, 1994.
- Lersundi A, Mankin H, Mourikis A, et al: Chondromyxoid fi broma—a rarely encountered and puzzling tumor, Clin Orthop Relat Res 439:171, 2005.
- Macdonald D, Fornasier V, Holtby R: Chondromyxoid fi broma of the acromion with soft tissue extension: case report, Skeletal Radiol 29:168, 2000.
- Marin C, Gallego C, Manjon P, et al: Juxtacortical chondromyxoid fi broma: imaging fi ndings in three cases and a review of the literature, Skeletal Radiol 26:642, 1997.
- McGrory BJ, Inwards CY, McLeod RA, et al: Mayo Clinic tumor rounds: chondromyxoid fi broma, Orthopedics 18:307, 1995.
- Mitchell M, Sartoris DJ, Resnick D: Case report 713, Skeletal Radiol 21:252, 1992.
- Nimityongskul P, Anderson LD, Dowling EA: Chondromyxoid fi broma, Orthop Rev 21:863, 1992.
- O’Connor PJ, Gibbon WW, Hardy G, et al: Chondromyxoid fi broma of the foot, Skeletal Radiol 25:143, 1996.
- Simon MA, Springfi eld D, eds: Surgery for bone and soft-tissue tumors, Philadelphia, 1998, Lippincott-Raven. Troncoso A, Ro JY, Edeiken J, et al: Case report 798, Skeletal Radiol 22:445, 1993.
- Tsuchiya H, Tomita K, Tsuchida T, et al: Case report 741, Skeletal Radiol 21:339, 1992.
- Unni KK: Dahlin’s bone tumors: general aspects and data on 11,087 cases, 5th ed, Philadelphia, 1996, Lippincott-Raven. White PG, Saunders L, Orr W, et al: Chondromyxoid fi broma, Skeletal Radiol 25:79, 1996.
- [Wold LE, McLeod RA, Sim FH, et al: Atlas of orthopedic pathology, Philadelphia, 1990, Saunders.
Osteoblastoma Adle C: Multifocal osteoblastoma of the hand: case report, Skeletal Radiol 29:601, 2000.](https://pubmed.ncbi.nlm.nih.gov/?term=Wold%20LE%2C%20McLeod%20RA%2C%20Sim%20FH%2C%20et%20al%3A%20Atlas%20of%20orthopedic%20pathology%2C%20Philadelphia%2C%201990%2C%20Saunders.%0A%0AOsteoblastoma%20Adle%20C%3A%20Multifocal%20osteoblastoma%20of%20the%20hand%3A%20case%20report%2C%20Skeletal%20Radiol%2029%3A601%2C%202000.)
- Boriani S, Capanna R, Donati D, et al: Osteoblastoma of the spine, Clin Orthop Relat Res 278:37, 1992.
- Bullough PG: Bullough and Vigorita’s orthopaedic pathology, ed 3, London, 1997, Mosby-Wolfe. Castello JR, Garro L, San Miguel P, et al: Osteoblastoma of the scaphoid—long-term results following curettage: a case report, J Hand Surg 21A:426, 1996.
- Dickson KF, Johnston JO, Delgado E, et al: Case reports: osteoblastoma in the metaphysis of the distal femur, Orthopedics 16:903, 1993.
- Dransfeld JW, Resnik CS, Aisner SC, et al: Osteoblastoma in the fi bula, Skeletal Radiol 24:74, 1995.
- Forest M, Tomeno B, Vanel D, eds: Orthopedic surgical pathology: diagnosis of tumors and pseudotumoral lesions of bones and joints, Edinburgh, 1998, Churchill Livingstone. Frassica FJ, Waltrip RL, Sponseller PD, et al: Clinicopathologic features and treatment of osteoid osteoma and osteoblastoma in children and adolescents, Pediatr Orthop Oncol 127:559, 1996.
- Gil-Albarova J, Amillo S: Osteoblastoma causing rigidity of the elbow: case report, Acta Orthop Scand 62:602, 1991.
- Kenan S, Abdelwahab IF, Hermann G, et al: Osteoblastoma of the humerus associated with type I Gaucher’s disease: a case report, J Bone Joint Surg 78B:702, 1996.
- Nakatani T, Yamamoto T, Akisue T, et al: Periosteal osteoblastoma of the distal femur: case report, Skeletal Radiol 33:107, 2004.
- O’Connell JX, Rosenthal DI, Mankin HJ, et al: A unique multifocal osteoblastoma-like tumor of the bones of a single lower extremity: report of a case, J Bone Joint Surg 75A:597, 1993.
- Ozaki T, Liljenqvist U, Hillman A, et al: Osteoid osteoma and osteoblastoma of the spine: experiences with 22 patients, Clin Orthop Relat Res 397:394, 2002.
- Papagelopoulos PJ, Galanis EC, Sim FH, et al: Mayo clinic tumor rounds: osteoblastoma of the acetabulum, Orthopedics 21:355, 1998.
- Papagelopoulos PJ, Galanis EC, Sim FH, et al: Clinicopathologic features, diagnosis, and treatment of osteoblastoma, Orthopedics 22:244, 1999.
- Ruggieri P, McLeod RA, Unni KK, et al: Mayo Clinic tumor rounds: osteoblastoma, Orthopedics 19:621, 1996.
- Saifuddin A, Sherazi Z, Shaikah MI, et al: Spinal osteoblastoma: relationship between paravertebral muscle abnormalities and scoliosis, Skeletal Radiol 25:531, 1996.
- Saifuddin A, White J, Sherazi Z, et al: Osteoid osteoma and osteoblastoma of the spine: factors associated with the presence of scoliosis, Spine 23:47, 1998.
- Simon MA, Springfi eld D, eds: Surgery for bone and soft-tissue tumors, Philadelphia, 1998, Lippincott-Raven. Sovio OM, Beauchamp RD, Morton KS, et al: Osteoblastoma in the very young: report of two cases, J Pediatr Orthop 8:342, 1988.
- Unni KK: Dahlin’s bone tumors: general aspects and data on 11,087 cases, 5th ed, Philadelphia, 1996, Lippincott-Raven. Watanabe M, Kihara Y, Matsuda Y, et al: Benign osteoblastoma in the vertebral body of the thoracic spine: a case report, Spine 17:1432, 1992.
- [Wold LE, McLeod RA, Sim FH, et al: Atlas of orthopedic pathology, Philadelphia, 1990, Saunders.
Langerhans Cell Histiocytosis Beltran J, Aparisi F, Bonmati LM, et al: Eosinophilic granuloma: MRI manifestations, Skeletal Radiol 22:157, 1993.](https://pubmed.ncbi.nlm.nih.gov/?term=Wold%20LE%2C%20McLeod%20RA%2C%20Sim%20FH%2C%20et%20al%3A%20Atlas%20of%20orthopedic%20pathology%2C%20Philadelphia%2C%201990%2C%20Saunders.%0A%0ALangerhans%20Cell%20Histiocytosis%20Beltran%20J%2C%20Aparisi%20F%2C%20Bonmati%20LM%2C%20et%20al%3A%20Eosinophilic%20granuloma%3A%20MRI%20manifestations%2C%20Skeletal%20Radiol%2022%3A157%2C%201993.)
- Bullough PG: Bullough and Vigorita’s orthopaedic pathology, 3rd ed, London, 1997, Mosby-Wolfe. Forest M, Tomeno B, Vanel D, eds: Orthopedic surgical pathology: diagnosis of tumors and pseudotumoral lesions of bones and joints, Edinburgh, 1998, Churchill Livingstone. Garg S, Mehta S, Dormans J: An atypical presentation of Langerhans cell histiocytosis of the cervical spine in a child, Spine 28:E445, 2003.
- Garg S, Mehta S, Dormans J: Langerhans cell histiocytosis of the spine in children: long-term follow-up, J Bone Joint Surg 86A:1740, 2004.
- Ghanem I, Tolo V, Ambra P, et al: Langerhans cell histiocytosis of bone in children and adolescents, J Pediatr Orthop 23:124, 2003.
- Giona F, Caruso R, Testi AM, et al: Langerhans’ cell histiocytosis in adults: a clinical and therapeutic analysis of 11 patients from a single institution, Cancer 80:1786, 1997.
- Henck ME, Simpson EL, Ochs RH, et al: Extraskeletal soft tissue masses of Langerhans’ cell histiocytosis, Skeletal Radiol 25:409, 1996.
- Howarth DM, Gilchrist GS, Mullan BP, et al: Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome, Cancer 85:2278, 1999.
- Islinger RB, Kuklo TR, Owens BD, et al: Langerhans’ cell histiocytosis in patients older than 21 years, Clin Orthop Relat Res 379:231, 2000.
- Levine SE, Dormans JP, Meyer JS, et al: Langerhans’ cell histiocytosis of the spine in children, Clin Orthop Relat Res 323:288, 1996.
- Lieberman PH, Jones CR, Steinman RM, et al: Langerhans cell (eosinophilic) granulomatosis: a clinicopathologic study encompassing 50 years, Am J Surg Pathol 20:519, 1996.
- Raab P, Hohmann F, Kuhl J, Krauspe R: Vertebral remodeling in eosinophilic granuloma of the spine: a long-term follow-up, Spine 23:1351, 1998.
- Raney RB Jr: Chemotherapy for children with aggressive fi bromatosis and Langerhans’ cell histiocytosis, Clin Orthop Relat Res 262:58, 1991.
- Sessa S, Sommelet D, Lascombes P, et al: Treatment of Langerhans-cell histiocytosis in children: experience at the Children’s Hospital of Nancy, J Bone Joint Surg 76A:1513, 1994.
- Simon MA, Springfi eld D, eds: Surgery for bone and soft-tissue tumors, Philadelphia, 1998, Lippincott-Raven. Tan G, Samson I, De Wever I, et al: Langerhans cell histiocytosis of the cervical spine: a single institution experience in four patients, J Pediatr Orthop 13B:123, 2004.
- Unni KK: Dahlin’s bone tumors: general aspects and data on 11,087 cases, 5th ed, Philadelphia, 1996, Lippincott-Raven. VanderWilde RS, Wold LE, McLeod RA, et al: Mayo Clinic tumor rounds: eosinophilic granuloma, Orthopedics 13:1301, 1990.
- Wold LE, McLeod RA, Sim FH, et al: Atlas of orthopedic pathology, Philadelphia, 1990, Saunders. Yasko AW, Fanning CV, Ayala AG, et al: Percutaneous techniques for the diagnosis and treatment of localized Langerhanscell histiocytosis (eosinophilic granuloma of bone), J Bone Joint Surg 80A:219, 1998.
- Yeom JS, Lee CK, Shin HY, et al: Langerhans’ cell histiocytosis of the spine: analysis of twenty-three cases, Spine 24:1740, 1999.
You Might Also Like
