Fibrous Lesions of Bone: Comprehensive Surgical Management
Key Takeaway
Fibrous lesions of bone encompass a diverse spectrum of developmental anomalies and true neoplasms, ranging from self-limiting nonossifying fibromas to locally aggressive desmoplastic fibromas. Management dictates a nuanced understanding of biomechanics, radiographic interpretation, and precise surgical indications. This comprehensive guide details the pathophysiology, step-by-step surgical approaches, and postoperative protocols for treating fibrous and cystic bone lesions in orthopedic practice.
INTRODUCTION TO FIBROUS LESIONS OF BONE
Fibrous lesions of bone represent a heterogeneous group of developmental anomalies, reactive processes, and true neoplasms characterized by the replacement of normal cancellous or cortical bone with fibrous connective tissue. Encountered frequently in both pediatric and adult orthopedic oncology, these lesions range from entirely benign, self-limiting entities to locally aggressive tumors requiring wide surgical resection.
A profound understanding of the biomechanical implications of these lesions, their natural history, and their radiographic signatures is paramount for the practicing orthopedic surgeon. Misdiagnosis or inappropriate surgical intervention can lead to catastrophic structural failure, unnecessary morbidity, or high rates of local recurrence.
NONOSSIFYING FIBROMA (NOF)
Nonossifying fibromas—also referred to in the literature as metaphyseal fibrous defects, fibrous cortical defects, or fibroxanthomas—are the most common developmental abnormalities of the pediatric skeleton. Current epidemiological data suggest they occur in up to 35% of all growing children.
Pathophysiology and Clinical Presentation
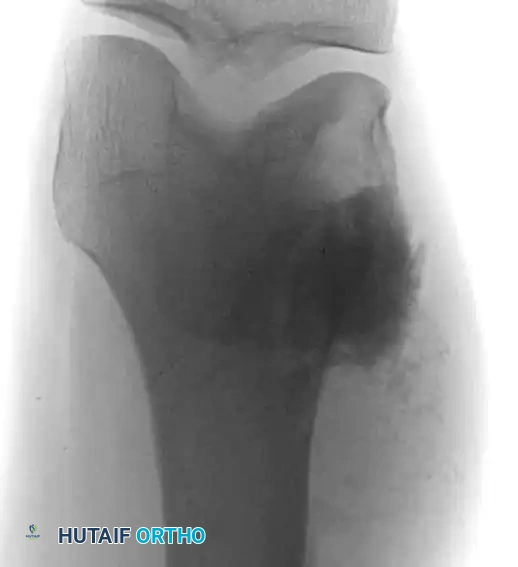
Typically discovered incidentally, these lesions manifest in the metaphyseal regions of long bones in patients aged 2 to 20 years. The distribution is highly predictable: approximately 40% occur in the distal femur, 40% in the proximal or distal tibia, and 10% in the fibula.
Histologically, the defect is densely packed with spindle-shaped fibroblasts arranged in a characteristic whorled or "storiform" pattern. High cellularity is accompanied by the almost ubiquitous presence of multinucleated giant cells and lipid-laden foam cells.
Radiographic Evaluation
On plain radiography, an NOF presents as a well-defined, lobulated, and eccentrically located radiolucency within the metaphysis.
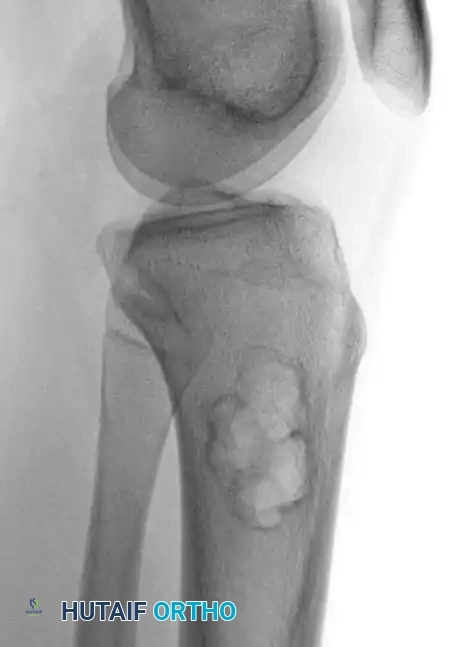
Fig. 20-8 A and B: Anteroposterior and lateral radiographs demonstrating a classic nonossifying fibroma of the proximal tibia in a 15-year-old patient. Note the eccentric location and sclerotic margins.
A multilocular appearance with distinct ridges in the bony wall, sclerotic scalloped borders, and focal erosion of the inner cortex are hallmark findings. Crucially, there is an absolute absence of periosteal reaction unless a pathological fracture has occurred.
Biomechanics and Surgical Indications
The vast majority of NOFs are asymptomatic and undergo spontaneous regression and ossification as the patient reaches skeletal maturity. However, surgical intervention is indicated under specific biomechanical conditions:
1. Impending Pathological Fracture: Lesions exceeding 50% of the transverse diameter of the host bone create a significant stress riser, drastically reducing the bone's torsional and bending strength.
2. Symptomatic Lesions: Pain in the absence of fracture suggests micro-trabecular failure or an alternative, more aggressive diagnosis.
3. Repeated Trauma: High-demand athletes with large lesions may require prophylactic stabilization.
Surgical Warning: The "50% rule" is a widely taught heuristic, but biomechanical studies indicate that the length of the lesion relative to the bone diameter and its specific location (e.g., high-stress areas like the distal medial femur) must also dictate the decision for prophylactic fixation.
Operative Technique: Extended Curettage and Bone Grafting
When surgery is indicated, extended curettage with bone grafting is the gold standard. Recurrence following meticulous curettage is exceptionally rare.
- Positioning and Setup: The patient is positioned supine on a radiolucent Jackson table. Fluoroscopy is utilized to localize the lesion precisely.
- Surgical Approach: A direct approach over the eccentric lesion is performed, carefully retracting neurovascular structures.
- Cortical Windowing: An oval or oblong cortical window is created using a high-speed burr. Sharp corners must be avoided to prevent the creation of postoperative stress risers.
- Curettage: Aggressive intralesional curettage is performed using angled curettes to reach all lobulated recesses. The cavity is then expanded using a high-speed spherical burr until healthy, bleeding punctate cortical bone is visualized (the "paprika sign").
- Adjuvant Treatment: Pulsatile lavage is used to clear debris. Chemical adjuvants (e.g., hydrogen peroxide or phenol) may be applied to denature residual microscopic cells.
- Grafting: The defect is tightly packed with cancellous allograft, autograft, or a synthetic bone graft substitute. If the cortical defect is massive, prophylactic internal fixation (e.g., a neutralization plate) is applied to protect the construct during incorporation.
CORTICAL DESMOID
A cortical desmoid is a benign, self-limiting irregularity typically found on the posteromedial aspect of the distal femoral metaphysis. It is predominantly observed in active boys between the ages of 10 and 15 years.
Etiology and Diagnosis
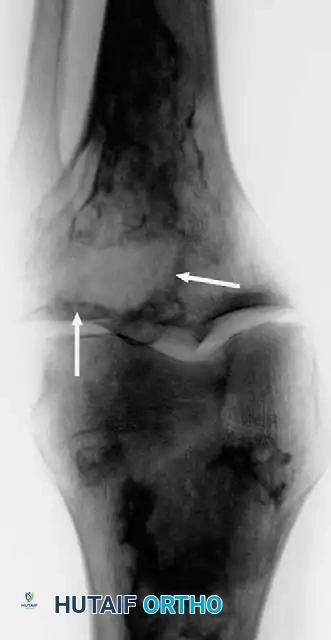
Current consensus attributes the cortical desmoid to a reactive, avulsive process secondary to repetitive traction from the aponeurotic insertion of the adductor magnus muscle. Clinical symptoms are rare but may include localized soft-tissue swelling and mild pain following exertion.
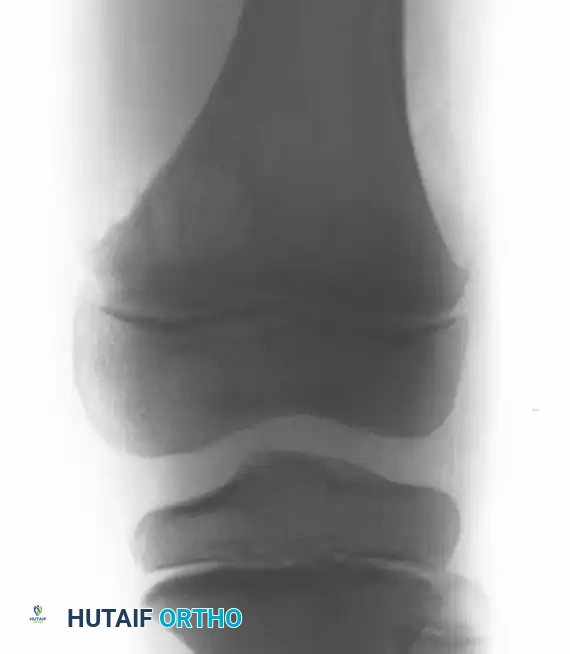
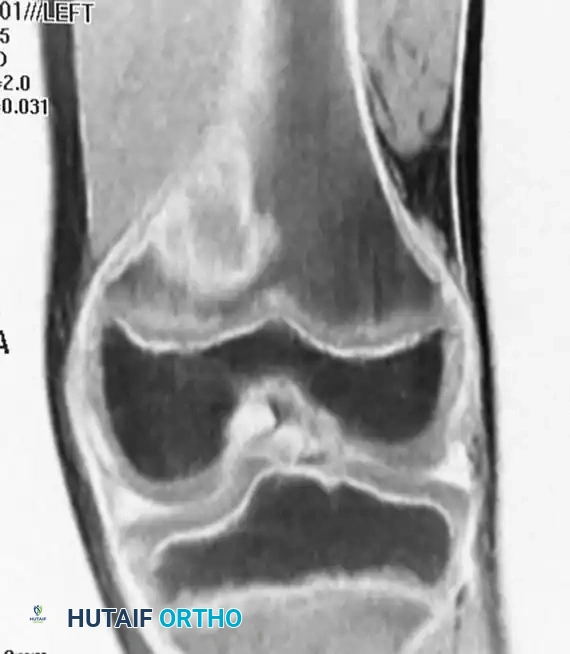
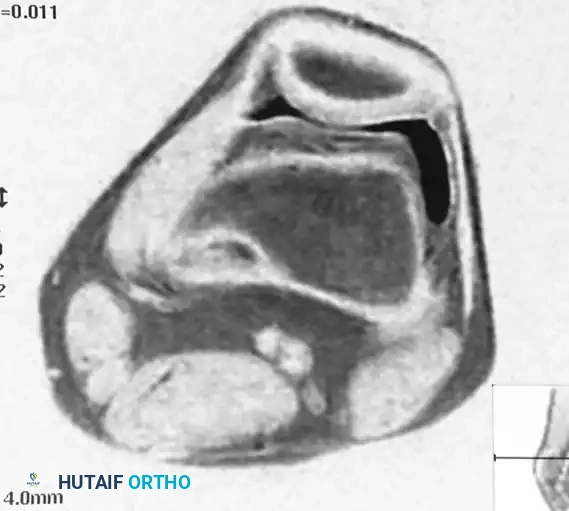
Fig. 20-9 A, B, and C: Eight-year-old boy with a cortical desmoid of the left femur. Anteroposterior radiograph (A) and corresponding Coronal (B) and Axial (C) MRI scans revealing cortical erosion with a sclerotic base.
The lesion is optimally visualized on an oblique radiograph with the lower extremity externally rotated 20 to 45 degrees. Advanced imaging (MRI) demonstrates focal cortical erosion with a thickened, sclerotic base and adjacent soft-tissue edema.
Clinical Pearl: The cortical desmoid is a classic "do not touch" lesion. Because its radiographic and MRI appearance can mimic early osteosarcoma or periosteal reactions, recognizing its exact posteromedial location is critical. Biopsy is strictly contraindicated, as the reactive histological features can be misinterpreted as a malignancy. No treatment is necessary.
BENIGN FIBROUS HISTIOCYTOMA (BFH)
Benign fibrous histiocytoma of bone is a rare entity, first delineated by Dahlin in 1978. While histologically indistinguishable from a nonossifying fibroma, its biological behavior and clinical presentation are markedly different.
Distinguishing Features
Unlike NOFs, which are developmental and self-limiting, BFH is considered a true neoplasm. Key differentiating factors include:
* Age: BFH typically presents in older adults, usually between 30 and 40 years of age.
* Location: While NOFs are strictly metaphyseal, BFH frequently involves the diaphysis, epiphysis, or the axial skeleton (pelvis).
* Radiography: BFH presents as a well-defined, lytic, and expansile lesion, often with minimal periosteal reaction. Bone scintigraphy (bone scans) will show mild to moderate increased radiotracer uptake.
Surgical Management
Because BFH exhibits a much more aggressive biological behavior and a high propensity for local recurrence, simple curettage is inadequate.
* Extended Curettage: Aggressive burring with the use of local adjuvants (phenol, liquid nitrogen, or argon beam coagulation) is mandatory.
* Wide Resection: In cases of massive bone destruction or recurrence, wide en bloc resection with structural reconstruction (megaprosthesis or structural allograft) is recommended.
FIBROUS DYSPLASIA
Fibrous dysplasia is a profound developmental anomaly of osteogenesis where normal bone and marrow are replaced by structurally inferior fibrous tissue and immature, woven spicules of bone. It exists in both monostotic (single bone) and polyostotic (multiple bones) forms.
Pathophysiology and Syndromic Associations
The disease is driven by a post-zygotic activating mutation in the GNAS1 gene, leading to overproduction of cAMP and subsequent abnormal osteoblast differentiation.
Fibrous dysplasia may present as part of a broader syndromic complex:
* McCune-Albright Syndrome: Polyostotic fibrous dysplasia, irregular cutaneous pigmentation (café-au-lait spots with "coast of Maine" borders), and precocious puberty or other endocrine hyperfunction.
* Mazabraud Syndrome: Polyostotic fibrous dysplasia associated with intramuscular myxomas.
Malignant transformation (typically to osteosarcoma or fibrosarcoma) is rare but documented, particularly in patients who have previously received radiation therapy—making radiotherapy strictly contraindicated.
Radiographic and Histological Evaluation
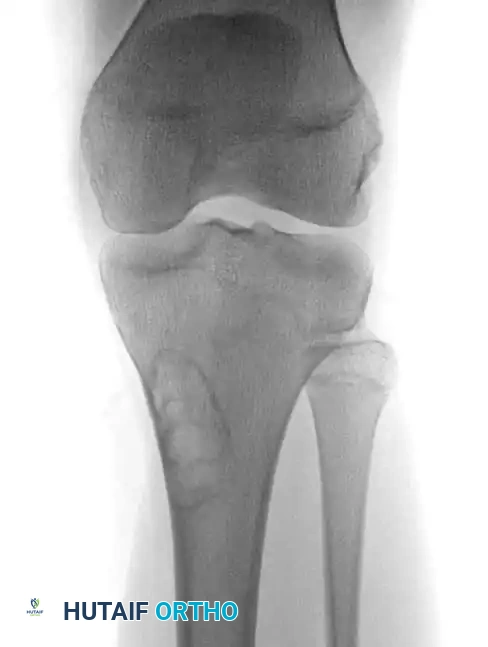
The radiographic signature of fibrous dysplasia is a radiolucent, expansile lesion with a characteristic granular, "ground-glass" matrix.
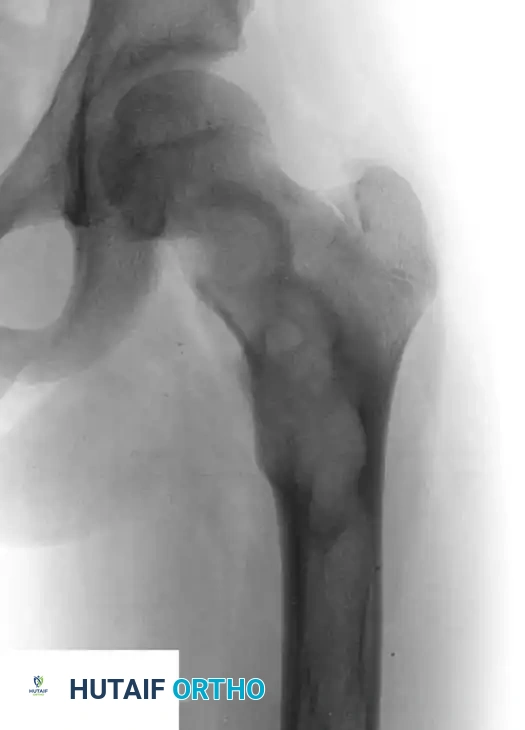
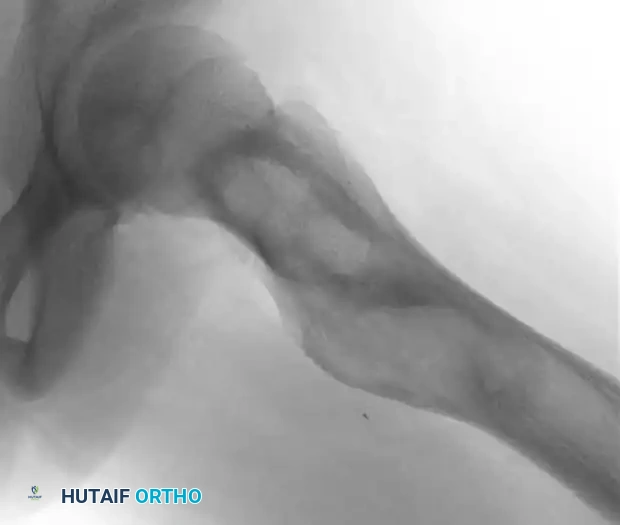
Fig. 20-10 A and B: Left hip of a 12-year-old girl demonstrating the classic ground-glass appearance and expansile nature of fibrous dysplasia.
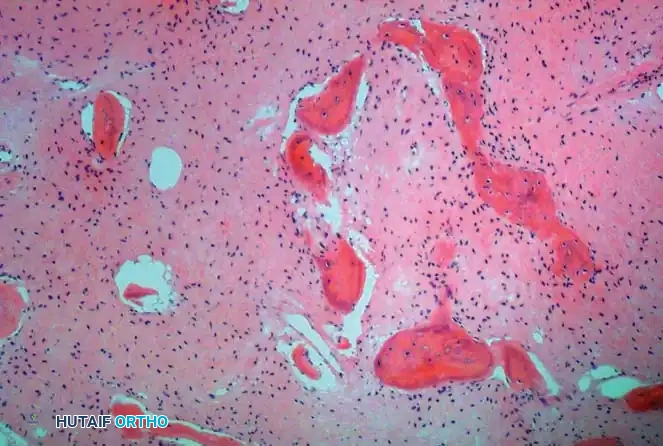
Fig. 20-11: Typical histological appearance of fibrous dysplasia, showing irregular, woven bone spicules ("Chinese character" shapes) embedded in a bland fibrous stroma without osteoblastic rimming.
Surgical Management and Biomechanics
Surgical intervention is indicated for significant pain, progressive deformity (such as the classic "shepherd's crook" deformity of the proximal femur), or actual/impending pathological fractures.
- Internal Fixation: Due to the structurally deficient nature of the dysplastic bone, load-sharing devices (intramedullary nails) are vastly superior to load-bearing devices (plates). Intramedullary fixation spans the entire bone, protecting it from future fractures.
- Bone Grafting Principles:
> Surgical Warning: Cancellous bone autograft or allograft is rapidly resorbed and replaced by the dysplastic host tissue. Therefore, cortical bone grafts (strut allografts) are strictly preferred due to their slow resorption profile and superior structural support. - Medical Management: Intravenous bisphosphonates (e.g., pamidronate or zoledronic acid) have shown significant efficacy in reducing bone pain and decreasing the rate of osteolysis in extensive polyostotic disease.
OSTEOFIBROUS DYSPLASIA (CAMPANACCI DISEASE)
Osteofibrous dysplasia is a rare, benign fibro-osseous lesion that almost exclusively affects the tibia and fibula. It typically presents in the first two decades of life.
Clinical Presentation and Diagnosis
The lesion is usually located in the diaphyseal or diaphyseal-metaphyseal junction of the middle third of the tibia. Clinically, it presents as a painless enlargement and anterolateral bowing of the tibia. Pain is only present if a microfracture or complete pathological fracture occurs.
Radiographically, it appears as an eccentric, intracortical, osteolytic lesion that expands the anterior cortex.
Histology and Differential Diagnosis
Histologically, osteofibrous dysplasia exhibits a unique "zonal architecture": the center consists of loose, immature fibrous tissue, while the periphery features a band of bony trabeculae rimmed by active osteoblasts.
* Critical Differential: It is imperative to distinguish osteofibrous dysplasia from adamantinoma, a low-grade malignant bone tumor that occurs in the exact same location. Immunohistochemistry is vital; adamantinoma will stain positive for epithelial markers (cytokeratin), whereas osteofibrous dysplasia will not.
Surgical Strategy
The natural history is highly unpredictable. While some lesions regress spontaneously, most progress during childhood and stabilize after puberty.
* Skeletally Immature Patients: Recurrence rates after curettage or marginal resection are unacceptably high. Therefore, surgery is delayed if possible. Pathological fractures are treated nonoperatively in casts.
* Skeletally Mature Patients: Recurrence rates drop significantly. Surgical management is primarily aimed at correcting the anterolateral bowing deformity via corrective osteotomies and stable internal fixation.
DESMOPLASTIC FIBROMA
Desmoplastic fibroma is an extremely rare, locally aggressive benign bone tumor. It is the intraosseous counterpart to the soft-tissue desmoid tumor (aggressive fibromatosis).
Pathology and Presentation
Occurring most frequently in the second and third decades of life, it targets long tubular bones, the mandible, pelvis, and spine. Patients typically present with deep, unrelenting pain.
Radiographs reveal a well-circumscribed, lytic lesion with a narrow zone of transition, often featuring a thin rim of reactive bone and internal septations. Cortical destruction is common. MRI demonstrates low signal intensity on both T1- and T2-weighted images due to the dense collagenous matrix.
Grossly, the tissue is dense, tough, and rubbery. Microscopically, it is hypocellular, heavily collagenized, and lacks nuclear atypia or frequent mitoses.
Surgical Resection
Desmoplastic fibroma does not metastasize, but its local invasiveness dictates aggressive management.
* Wide Resection: En bloc resection with negative margins is the definitive treatment of choice to prevent recurrence.
* Extended Curettage: In anatomically complex regions (e.g., the pelvis or spine) where wide resection would cause unacceptable morbidity, aggressive extended curettage with high-speed burring and chemical adjuvants may be attempted, though the patient must be counseled on the high risk of local recurrence.
* Adjuvant Therapies: While data is limited, anti-estrogen therapy (tamoxifen), NSAIDs, or low-dose chemotherapy may be considered for unresectable recurrences, mirroring soft-tissue desmoid protocols.
UNICAMERAL BONE CYSTS (UBC)
While technically cystic rather than purely fibrous, Unicameral Bone Cysts (Simple Bone Cysts) share similar metaphyseal locations and biomechanical risks, warranting inclusion in the differential management of benign pediatric bone lesions.
Pathogenesis
UBCs are fluid-filled cavities primarily found in the proximal humerus and proximal femur of growing children. They are classified as "active" when located within 1 cm of the physis and "latent" as they migrate diaphyseally with bone growth.
The prevailing pathophysiological theory suggests a focal defect in metaphyseal remodeling that blocks interstitial venous drainage. This localized venous hypertension leads to focal bone necrosis, fluid accumulation, and the concentration of bone-resorbing factors (prostaglandins, interleukins, and metalloproteinases). The cyst is lined by a thin (<1 mm) fibrous membrane composed of fibroblasts and osteoclast-like giant cells.
Surgical Management Protocols
Small, asymptomatic lesions in the upper extremity are managed with serial radiographic observation. Intervention is required for large lesions at high risk of fracture, or lesions in weight-bearing bones (lower extremity).
- Conservative Management of Fractures: Pathological fractures in the upper extremity are often treated in a sling or cast. The fracture hematoma occasionally initiates spontaneous cyst "healing."
- Intramedullary Nailing: For the proximal femur or humerus, flexible intramedullary nailing (e.g., Nancy nails or titanium elastic nails) provides immediate biomechanical stability, decompresses the cyst continuously, and allows early mobilization without casting.
- Curettage and Grafting: Formal curettage with bone grafting is reserved for recalcitrant cysts or proximal femur lesions requiring rigid internal fixation (e.g., pediatric dynamic hip screws).
Step-by-Step Steroid Injection Technique
Introduced in the 1970s to circumvent the 50% recurrence rate of simple curettage, intralesional steroid injection remains a frontline, minimally invasive treatment.
- Anesthesia and Positioning: The procedure is performed under deep sedation or general anesthesia in the operating room.
- Needle Placement: Under strict fluoroscopic guidance, a heavy 18-gauge spinal needle is advanced through the overlying cortex into the dependent portion of the cyst. A second 18-gauge needle is placed at the opposite (superior) end to act as a vent.
- Fluid Aspiration: The diagnosis is confirmed by the efflux of clear, straw-colored serous fluid. If the fluid is bloody, a telangiectatic osteosarcoma or aneurysmal bone cyst must be ruled out.
- Cystography: Radiopaque contrast dye is injected to confirm that the needles are within the cyst cavity and to identify any internal septations that might require separate injections.
- Injection: The cyst is thoroughly flushed with saline. Finally, 80 to 200 mg of methylprednisolone acetate (Depo-Medrol) is injected into the cavity.
- Mechanism of Action: The steroid acts by inhibiting the local prostaglandin-mediated bone resorption and decreasing the oncotic pressure of the cyst. Serial injections every 2 to 3 months may be required until radiographic consolidation is achieved.
You Might Also Like